Introduction
Somatic indel (insertion or deletion) mutations are frequently found in cancer genomes. Large-scale analysis from The Cancer Genome Atlas has revealed about 8300 unique somatic indels across roughly 4,000 cases for the 10 most common tumor types (including lung, breast, brain, and colorectal).1 Indels in genes such as BRCA1, BRCA2, and EGFR exon 20 that are involved in either DNA damage repair or activation of oncogenic pathways are well documented and serve as biomarkers for therapeutic interventions.
Bioinformatics tools play a critical role in accurately extracting these signals. These tools must be rigorously evaluated and optimized to accurately identify indel variants. To this end, the Food and Drug Administration (FDA) launched the NCTR Indel Calling from Oncopanel Sequencing Data Challenge2 to give the genomics community an opportunity to develop, validate, and benchmark somatic indel calling algorithms on oncopanel sequencing data sets.
Illumina participated in this challenge with the DRAGEN 4.0 somatic small variant caller.3 DRAGEN produced indel calls with the highest precision and overall accuracy in the applicability challenge (Panel X). This validates the accuracy of the DRAGEN algorithms in detecting somatic small variants and builds upon the previous DRAGEN win in the PrecisionFDA Truth Challenge V24 for germline small variant calling in the difficult-to-map regions.
In this post, we briefly describe the somatic indel challenge, summarize the results, and discuss aspects of the DRAGEN somatic indel methods that contributed to high accuracy variant calling.
PrecisionFDA challenge overview
As adoption of panel-based sequencing continues to grow in oncology clinical and translational laboratories, it is critical for the sequencing community to have access to high quality variant calling tools and gold standard data sets.5 The FDA initiated the Sequencing Quality Control Phase 2 project6 to develop standard analysis protocols and quality control metrics for the use of next-generation sequencing data, including oncopanel sequencing.
The oncopanel data for this challenge was created by the PrecisionFDA team from an artificial tumor sample created from equal mass mixture of ten Universal Human Reference RNA (UHRR) cancer cell-lines.7 Truth set indels were determined by three whole-exome sequencing (WES) runs of each cell line. A truth set for indels was manually curated by the challenge organizers using each individual cell-line data set. This truth (not shared with participants) was used for evaluation of different pipeline submissions.
The challenge comprised two phases (Figure 1): In phase 1, participants were provided raw sequencing data (FASTQs) from UHRR admixture DNA using two oncopanels known as Panel A (≈3.5 Mb) and Panel B (≈1 Mb). Each library was prepared in three different labs and sequenced four times to achieve a total of 12 sequencing replicates per panel. Participants were asked to process these data with their pipelines to optimize for the data set type and establish a well-defined indel calling pipeline. At the end of Phase 1, participants were asked to submit 12 variant calling results for both Panel A and Panel B.
Researchers with submissions in Phase 1 of this challenge were invited to participate in Phase 2, in which they were no longer permitted to modify their pipelines. A new data set (Panel X) was shared with them to evaluate the generalizability of each of the frozen pipelines. Panel X was intended to simulate real-world performance of indel calling pipelines: Frequently in clinical and translational laboratories, bioinformatics pipelines are optimized, their parameters locked, their performance validated; then they are run over many samples in a locked state. In both phases, the challenge focused only on indels of less than 50 bp in the targeted region shared by the organizers. For more details on the challenge, please visit its website here.
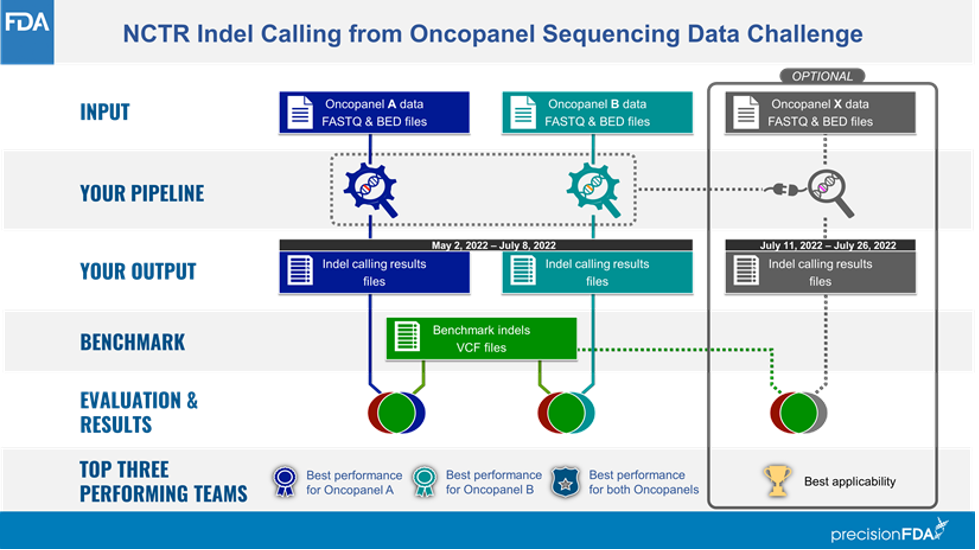
Source: Figure 1 at https://precision.fda.gov/challenges/21
PrecisionFDA challenge results
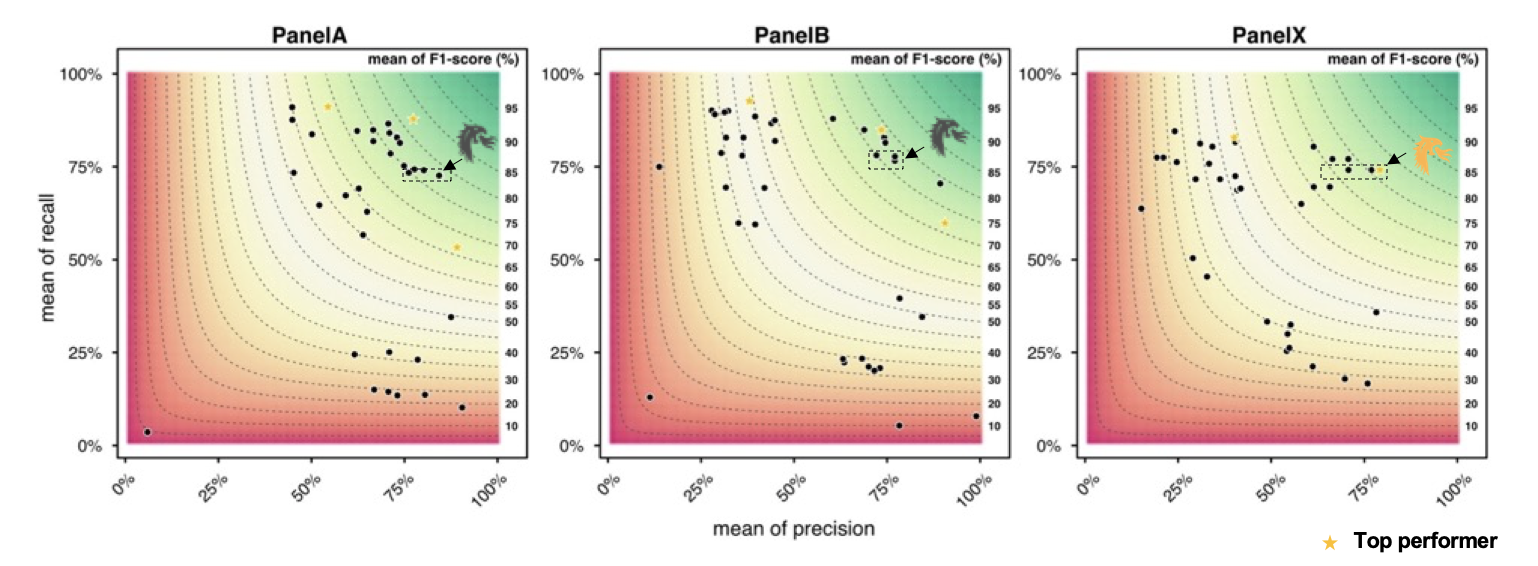
The selection criteria for the competition’s awards were announced once the challenge was completed. For each panel, the best performers in terms of the highest F1 score, highest precision, and highest recall were announced. An F1 score above the median of all submitters of that panel was required to place among the winning entries to prevent over-optimization of precision in favor of recall, and vice versa.
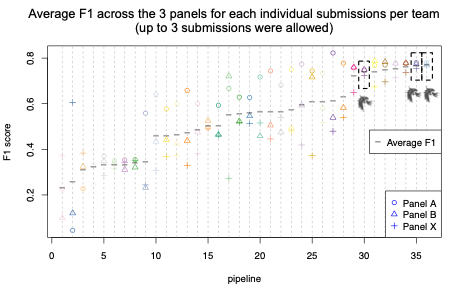
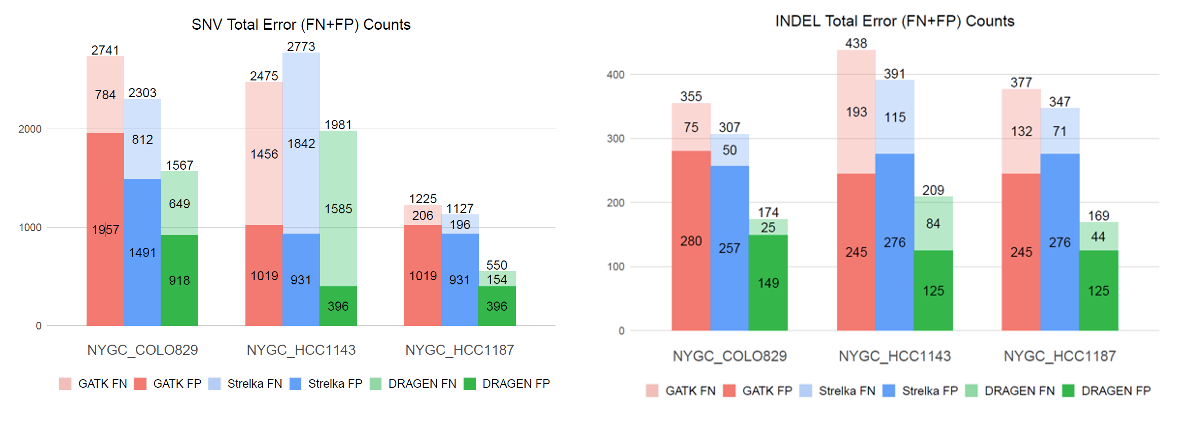
A reliable bioinformatics pipeline must have a low number of false negatives (FNs) and false positives (FPs). It is beneficial for a general bioinformatics pipeline to be consistently accurate across different data sets (that is, it must be generalizable). The DRAGEN submission achieved high precision, recall, and F1 score across all three panels with the best precision and F1 compared to all other participants in the applicability challenge (Panel X) (Figures 2–4). Moreover, DRAGEN showed highest accuracy when averaged across all panels and performance was consistent across the panels, highlighting that DRAGEN high performance is generalizable over multiple different panels using a single parameter set, avoiding the need to make panel-specific optimizations that are frequently required by other bioinformatics solutions (Figures 2–4).

Source: Figure 2 at https://precision.fda.gov/challenges/22/results
Source: Modified from Figure 1 at https://precision.fda.gov/challenges/22/results
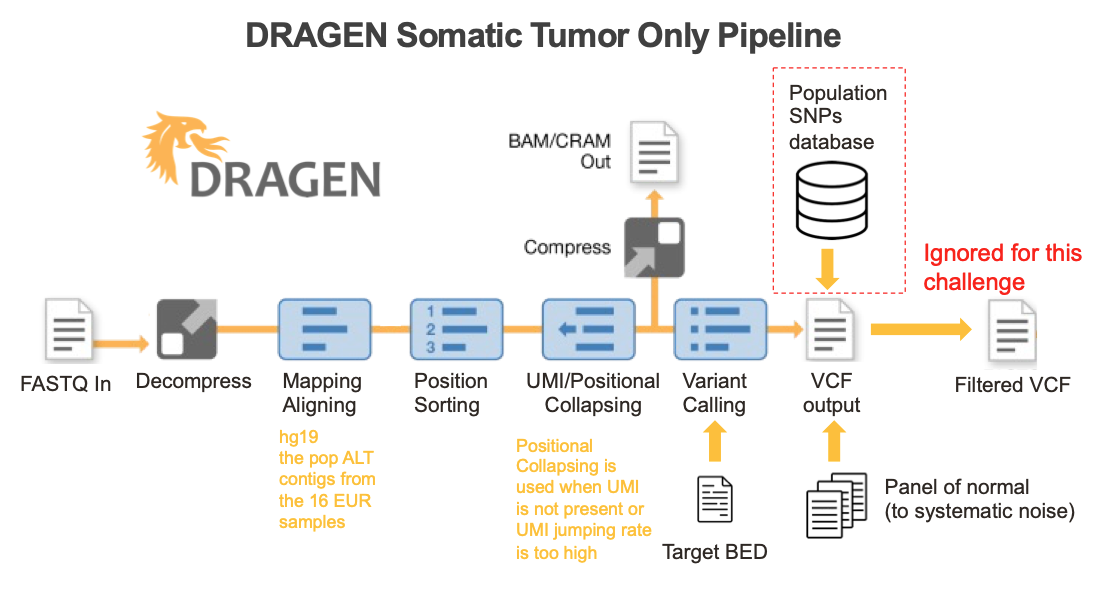
DRAGEN somatic workflow and methods
DRAGEN enables seamless end-to-end processing of raw sequencing data generated from any oncopanel
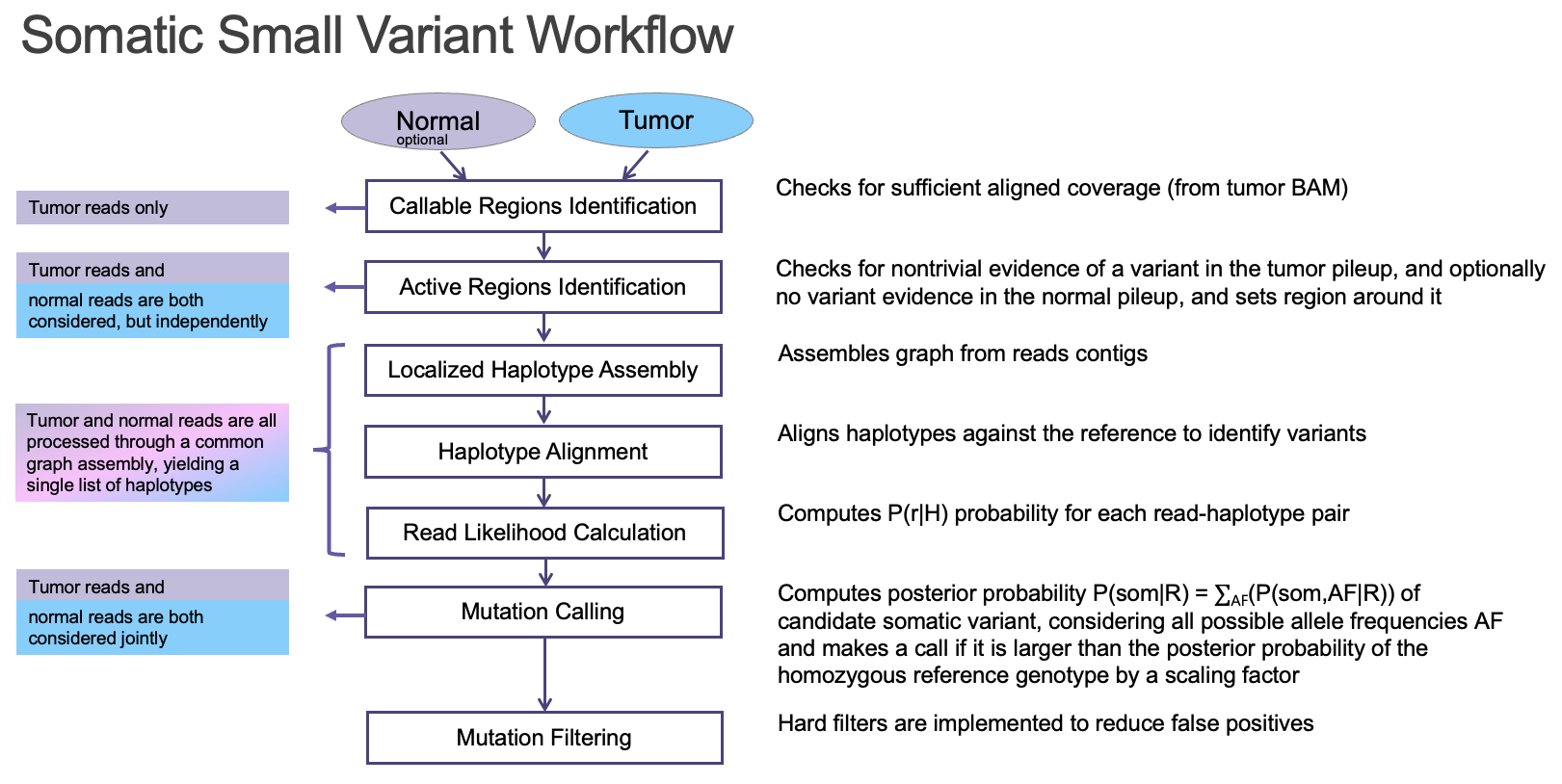
An ideal bioinformatics pipeline enables seamless end-to-end processing of any sequencing data types. To achieve this goal, DRAGEN offers a complete solution to align raw sequencing reads to reference genome, identify PCR duplicates, suppress sequencing errors in the presence or absence of unique molecular identifiers (UMIs), detect somatic indels, and provide a somatic quality score that can be leveraged to filter low confidence variants and reduce FPs (Figure 5).
Key features implemented in the DRAGEN somatic small variant calling pipeline include: 1) DRAGEN leverages improved alignment to a multi-genome (graph) reference, which is vital to achieving high accuracy in difficult-to-map regions of the genome; 2) DRAGEN facilitates processing of oncopanel data in the presence or absence of UMI sequence, making effective use of UMIs for noise correction when present while providing an optional alternative noise-reduction method if UMI barcodes are not used; 3) DRAGEN uses a single (highly accurate) variant caller, avoiding the need for computationally expensive and difficult-to-manage ensemble-based calling methods that incorporate multiple different open-source solutions. Together with field-programmable gate array (FPGA)–based hardware acceleration of several core algorithms in the aligner and variant caller, this results in the fastest available pipeline for processing any sequencing data with top accuracy.
DRAGEN multi-genome-based aligner enables highly accurate indel detection
A key feature of DRAGEN is its multi-genome-based aligner. As shown previously by the Illumina DRAGEN team,4 as well as others,8 leveraging a multi-genome reference can resolve ambiguous mapping of reads in difficult-to-map regions and therefore reduce alignment errors, yielding improved variant calling accuracy. The DRAGEN multi-genome reference contains population single-nucleotide polymorphisms (SNPs) and hundreds of alternate population haplotypes. Consequently, by providing the read mapper with information regarding an alternative haplotype path, ambiguously mapped reads can be recovered effectively. To facilitate building a multi-genome reference, DRAGEN 4.0+ is now shipped with a multi-genome-building toolkit, enabling researchers to seamlessly generate their own multi-genome reference of interest. The DRAGEN multi-genome aligner was enabled in the DRAGEN submissions to the challenge.
DRAGEN flexible architecture allows effective sequencing noise suppression with or without UMI tags using positional collapsing
UMIs are a set of “barcodes” introduced at the end of either one read (simplex) or both ends of paired end reads (duplex) to distinguish unique DNA or RNA molecules from PCR duplicates (Figure 6A). This well-established approach has been widely adopted to reduce sequencing noise, facilitating the discrimination of low allele frequency (AF) mutations from sequencing artefacts. After UMI collapsing, a high-quality consensus read is created, reducing error rate and improving variant calling accuracy, especially when a library is sequenced deeply (more than 2000 times).
Alternatively, the DRAGEN pipeline can process oncopanel sequencing data when UMI barcodes are not utilized. In this case, DRAGEN recruits a positional collapsing algorithm. Briefly, once reads are mapped to a reference genome, read families are generated based solely on alignment information and the sequence of the reads that start and end at the same location (Figure 6A). In this case, only variants that are supported by the majority of the reads in that family are considered true and the rest are ignored. This is in contrast with methods traditionally used to remove PCR duplicates, in that the latter cannot differentiate between noiseless and noisy reads that are mapped to the same start and end location—often accentuating error rate.
A comparison between sequencing error in in-house samples that were processed through a 1.94 Mb oncopanel (TruSight Oncology 500) demonstrates more than a twofold reduction in error rate when positional or UMI collapsing was applied, compared to the traditional remove duplicates method (also known as dedupping) (Figure 6B). Additionally, UMI and positional collapsing are seen to produce similar error rate reduction. We noted highly concordant small variant calling when positional collapsing (while ignoring UMIs) was compared to duplex UMI collapsing (Figure 6C).
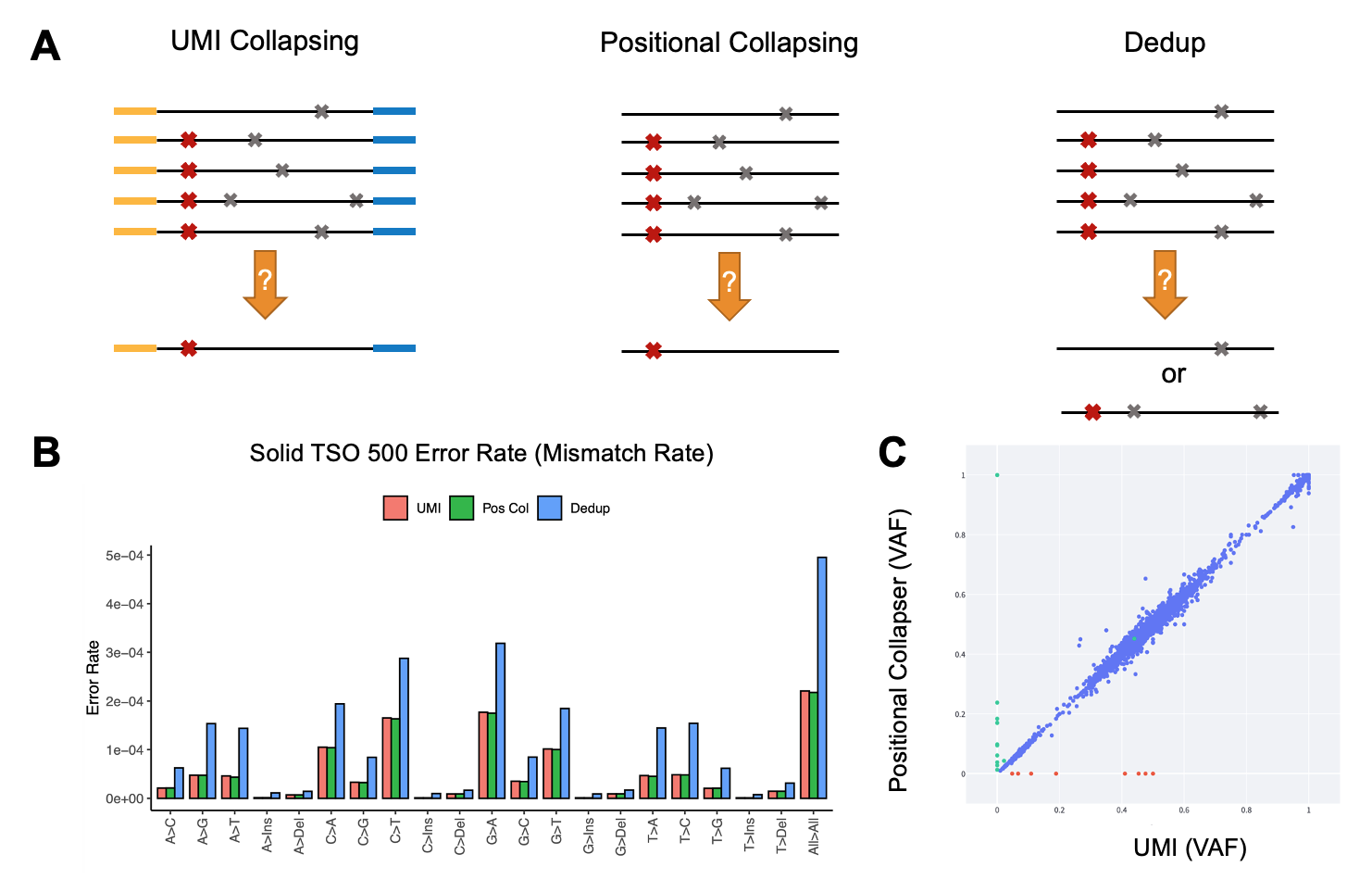
Figure 6A. Comparison between UMI collapsing, positional collapsing, and removing PCR duplicates. In the left panel, the yellow and blue regions of the reads represent duplex UMI sequences. Red and gray cross symbols show true mutation versus sequencing artifacts.
Figure 6B. Error rate (mismatch rate compared to reference) for the three methods of noise suppression. Data generated in house using samples that were sequenced by the TSO 500 oncopanel.
Figure 6C. DRAGEN variant calling using consensus reads generated by UMI collapsing versus positional collapsing. Data points represent AF of the detected mutations. Blue points represent concordant calls, while red and green points illustrate variants private to UMI versus positional collapsing only.
DRAGEN benefits from an improved haplotype caller and exceeds in accuracy compared to other well-established tools in identifying low AF somatic indels
The DRAGEN somatic variant caller9 combines the strengths of several trusted open-source algorithms with new DRAGEN-specific innovations. The overall architecture (Figure 7) is based on that of GATK 4.0, while the core Bayesian genotyping model (the “Mutation Calling” component in Figure 5A) is based on that of Strelka2, which performs joint analysis of tumor and normal samples in the case of a tumor/normal workflow. The DRAGEN team has added several improvements, including sample-specific estimation of indel error rates and nucleotide error bias (both of which inform the parameters for the hidden Markov model that performs read likelihood calculation), and probability models of common error modes such as strand bias, orientation bias, and mismapping. Together, these innovations result in accuracy that outperforms both of the above predecessor tools (Figure 8).
As described above, DRAGEN supports the processing of tumor samples in the presence or absence of a paired normal sample. A paired normal sample enables DRAGEN to more accurately remove germline variants. In its absence, DRAGEN can remove germline variants using 1) common SNPs in publicly available datasets (dbSNP or gnomAD, for example), and 2) the proximity of other germline variants (also known as proxy filter). Nonetheless, in this challenge, participants were instructed not to remove germline variants, since the majority of somatic true positives were present as germline variants in one or more cell lines prior to mixture.
The presence of a paired normal sample also improves accuracy by helping DRAGEN to characterize systematic sequencing noise that can result from PCR artifacts, polymerase slippage, formation of secondary structures such as G-quadruplex, and so on. Regardless of whether a normal sample is present (but especially important in its absence), the DRAGEN pipeline also allows users to generate a systematic noise file (also known as a panel of normals, or PON) to capture lab- and process-specific noise characteristics by estimating position-specific error rates from a panel of normal samples. These error rates are then used to filter calls at specific positions in the tumor sample if the signal at those positions can be statistically explained as noise.*
*Currently, most pipelines (including DRAGEN) do not differentiate between SNV and indels (of different size) when a PON is used. However, in this challenge, a new systematic noise approach was applied to account for SNV vs. INDEL errors. Briefly, a variant was filtered if AF of the exact allele is seen in any samples from PON was AF<30%. If a variant has AF>30% in an unpaired normal, the variant was kept (likely germline variant). Overall, in-house benchmarking data revealed that using a PON can reduce FPs up to 15% in oncopanel sequencing data.
Conclusion
This challenge represented an opportunity for the Illumina DRAGEN team to demonstrate the capabilities of the DRAGEN somatic indel calling pipeline. DRAGEN won top place in precision and F1 score in the applicability challenge (Panel X). DRAGEN also showed high and consistent accuracy across the three panels by earning the best F1 score when averaged across all three panels. This highlights that DRAGEN high performance in somatic indel calling is generalizable over multiple different panels using a single parameter set.
Acknowledgments
We would like to thank PrecisionFDA and all participants for their invaluable effort to organize and participate in this challenge. Similar to previous PrecisionFDA challenges, it provided a unique setting to evaluate and optimize bioinformatics pipelines. Furthermore, we thank all members of the DRAGEN team who have contributed to DRAGEN development in the past and during this challenge.
For Research Use Only. Not for use in diagnostic procedures.
References
1. GDC Data Portal with TCGA statistics
2. PrecisionFDA NCTR Indel Calling from Oncopanel Sequencing Data Challenge
4. DRAGEN Wins at PrecisionFDA Truth Challenge V2
5. Gong, B. et al. Cross-oncopanel study reveals high sensitivity and accuracy with overall analytical performance depending on genomic regions. Genome Biol 22, 109 (2021). https://doi.org/10.1186/s13059-021-02315-0
6. Sequencing benchmarked. Nat Biotechnol 39, 1027 (2021). https://doi.org/10.1038/s41587-021-01067-3
7. Jones, W. et al. A verified genomic reference sample for assessing performance of cancer panels detecting small variants of low allele frequency. Genome Biol 22, 111 (2021). https://doi.org/10.1186/s13059-021-02316-z
8. Ameur A: Goodbye reference, hello genome graphs. Nature biotechnology 2019, 37:866-868.
9. DRAGEN Somatic Small Variant Caller, Biorxiv, coming soon.