How to calculate NGS read length

What is Sequencing Read Length?
Next-generation sequencing (NGS) read length refers to the number of base pairs (bp) sequenced from a DNA fragment. After sequencing, the regions of overlap between reads are used to assemble and align the reads to a reference genome, reconstructing the full DNA sequence. Sequencing read lengths correspond directly to the sequencing reagents used on an NGS instrument—more chemistry cycles generate longer reads.

Why is NGS Read Length Important?
Choosing the right sequencing read length depends on your sample type, application, and coverage requirements. Because long reads allow for more sequence overlap, they are useful for de novo assembly and resolving repetitive areas of the genome with greater confidence. For other applications, such as expression profiling or counting studies, shorter reads are sufficient and more cost-effective than longer ones.
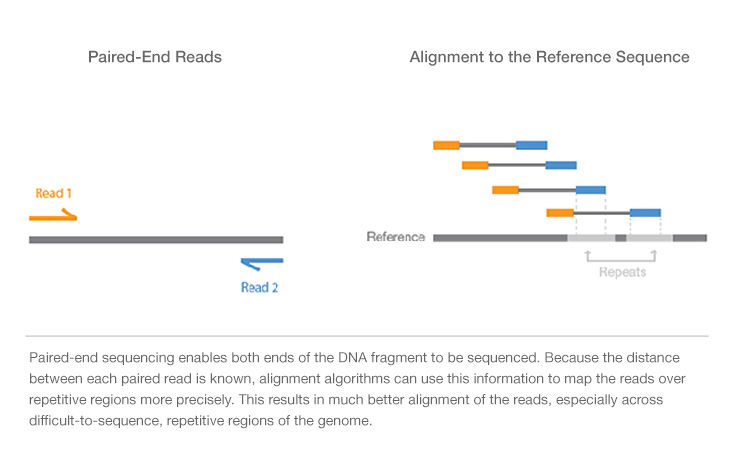
Sequencing Read Options
There are two sequencing read types: single-read and paired-end sequencing. Single-read sequencing involves sequencing DNA fragments from one end to the other. It is useful for some applications, such as small RNA sequencing, and can be a fast and economical option.
With paired-end sequencing, after a DNA fragment is read from one end, the process starts again in the other direction. In addition to producing twice the number of sequencing reads, this method enables more accurate read alignment and detection of structural rearrangements. Today, most researchers use the paired-end approach.
Learn MoreLibrary Prep Kit Selector
This tool provides recommended read lengths for different methods and Illumina library prep kits.
Use the ToolHow to Calculate Illumina Read Length
All Illumina sequencing reagents feature a certain number of sequencing cycles. These cycles are directly related to sequencing read length. Because one base is sequenced per cycle, the total number of cycles indicates the maximum number of bases that can be sequenced. You can use sequencing reagents to generate single continuous reads or for paired-end sequencing in both directions. (For example, a 300-cycle kit can be used for a 1 × 300 bp single-read run or a 2 × 150 bp paired-end run.)
| Application | Recommended Read Length |
|---|---|
| Whole-genome sequencing | 2 × 150 bp |
| Whole-exome sequencing | 2 × 150 bp |
| Targeted enrichment sequencing | 2 × 150 bp |
| Amplicon sequencing | Length of the entire amplicon insert |
| De novo sequencing | Ranges from 2 × 150 to 2 × 300 bp |
| Application | Recommended Read Length |
|---|---|
| Transcriptome analysis | 2 × 75 bp |
| Gene expression profiling | 1 × 50 bp |
| Small RNA sequencing | 1 × 50 bp |
Beginner's Guide to Next-Generation Sequencing
Considering bringing next-generation sequencing to your lab, but unsure where to start? These resources cover key topics in NGS and are designed to help you plan your first experiment.
Get Started
Sequencing Coverage Calculator
This tool helps you estimate sequencing coverage and contains read length recommendations for your experiment.
Use the ToolNGS Read Length and Coverage
Coverage depth refers to the average number of sequencing reads that align to, or "cover," each base in your sequenced sample. The Lander/Waterman equation1 is a method for calculating coverage (C) based on your read length (L), number of reads (N), and haploid genome length (G): C = LN / G
Considerations for RNA Sequencing Read Length
Different RNA-Seq experiment types have unique sequencing read length and depth requirements. This bulletin reviews read length and depth considerations and offers resources for planning experiments.
NGS Experimental Design & Protocols
Find tips and resources to help you plan your sequencing runs.
Learn MoreWhat is my maximum supported read length?
Depending on your sequencing platform and SBS kit version, your maximum supported read length can vary. Read the latest bulletin to learn more about your maximum read length along with other resources to optimize data quality.
Contact Us
Have questions about our products or how to get started with NGS? Connect with an Illumina representative.
Related Solutions
NGS Workflow Steps
The next-generation sequencing workflow contains three basic steps: library preparation, sequencing, and data analysis.
Sequencing Quality Scores
Sequencing quality scores measure the uncertainty of base calls, or the probability that a base is called incorrectly.
Planning Your NGS Budget
Learn more about the cost of NGS and how to budget for each step of the workflow.
Reference
- Lander ES, Waterman MS. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics. 1988;2(3):231-239.