Historically, detecting all these different sizes of variants has required using multiple different tests. By combining Illumina whole-genome sequencing (WGS) with secondary analysis algorithms built into the DRAGEN Bio-IT Platform, researchers can achieve high-sensitivity detection of all these different variant types using a mixture of methods. For the first time, DRAGEN v4.2 offers the option to support coverage-based copy number variant (CNV) calls with break-end structural variant (SV) results to better resolve small and medium genomic gain and loss events. This class of variant represents a great technical challenge, as it falls between classical sequencing and the array-based methods used for their smaller and larger brethren, respectively.
From a mathematical perspective, depth of coverage gets increasingly noisy for smaller event sizes (Figure 1) due to random fluctuations. For large events > 100 kb, the noise is hardly a factor at all. In the 10–100 kb range, the noise is present but typically not problematic. At the 1–10 kb scale, the noise is very high and the risk for false negative and false positive results is significant.
To address this noise issue, DRAGEN v4.2 jointly analyzes signals from the germline CNV and SV callers, identifies putative matches, updates annotations, filters, scores, and outputs the refined records. By leveraging junction signals from the SV caller and depth signals from the CNV caller, this approach allows for sensitive CNV detection down to 1 kb while also improving recall and precision across all length scales. This is achieved by rescuing previously low-quality calls if evidence is found from multiple signals, and by adjusting CNV break-ends to the more accurate SV break-ends.
WGS and DRAGEN bring together key features enabling high-powered copy number and structural variant analysis:
- Uniform coverage depth across the genome using PCR-free library prep allows for control-free normalization with a bin size of 1 kb
- Sequence data spanning introns and intergenic regions allows for the direct observation of breakpoint reads
- Algorithmic optimization using large datasets allows for tightly tuned filtering
- Synergy across coverage- and breakpoint-based callers allows for quality score boosting and event end refinement
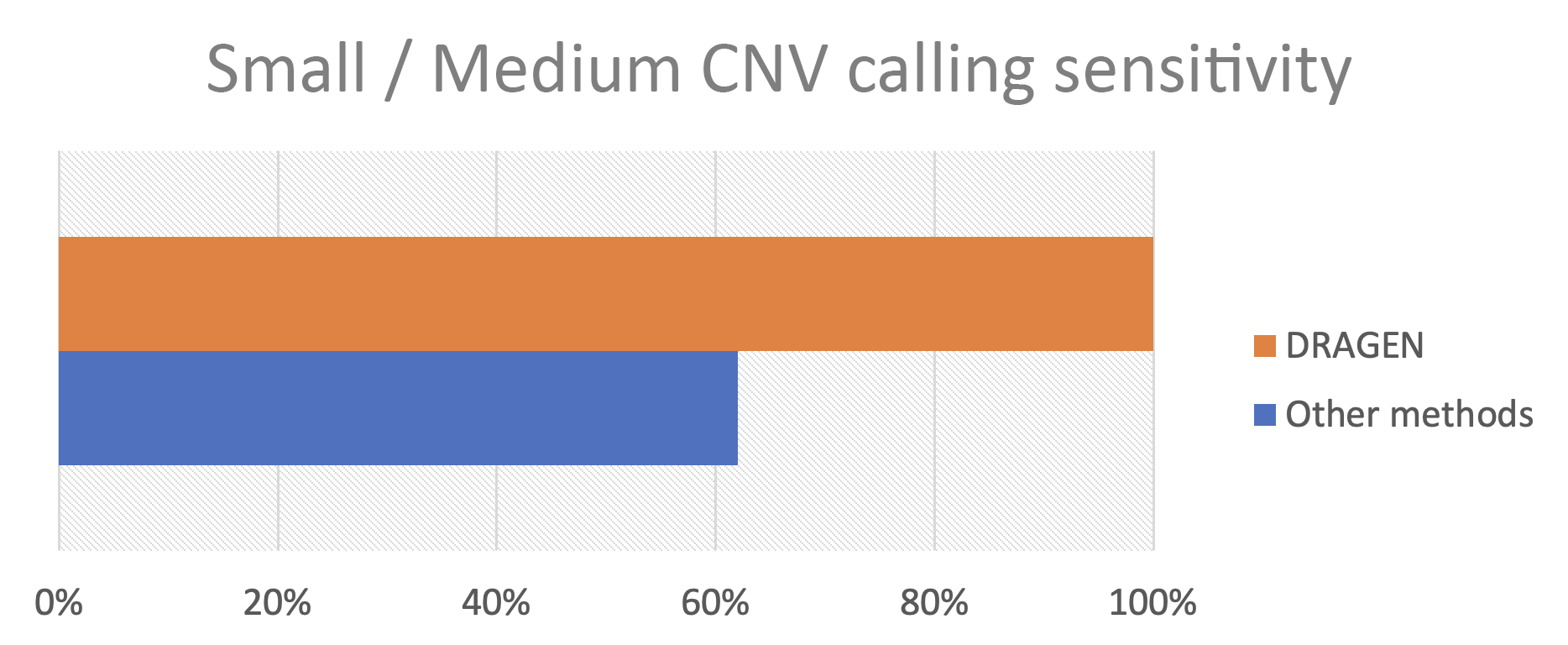
As summarized in work presented at the November 2023 Association for Molecular Pathology meeting, Francisco De La Vega, DSc, from Tempus Labs; Sean Irvine, PhD, from Real Time Genomics; and Sean Truong from Illumina led an effort to challenge CNV calling on small and medium CNV in medically relevant genes. We found that Illumina whole-genome sequencing combined with DRAGEN detected every variant thrown at it (Figure 2).
Researchers at the Broad Institute have also found that DRAGEN 4.2 demonstrates accurate CNV calling for events in this size range, especially for deletions between 5–10 kb.
These results tell a clear story: Genome sequencing is a highly sensitive platform for detection of small and medium CNV, a class of variation critical for research and medical applications such as hereditary cancer predisposition, cardiovascular disease, reproductive carrier screening, and more. This represents the culmination of years of effort by DRAGEN scientists and collaborators in the improvement of both coverage-based and break-end variant calling. We are excited about the possible applications our Illumina community will come up with using this updated technology.
Look out for future posts that go into more detail about additional uses of CNV and SV calling.