Abstract
In the summer of 2020, the PrecisionFDA Truth Challenge V2 invited genomics innovators to demonstrate their informatics workflows and show how they can improve DNA coverage and small variant calling accuracy in challenging genome regions. At that time, Illumina DRAGEN’s mapper + variant caller won the accuracy contest for Illumina reads in the Difficult-to-Map Regions and All Benchmark Regions (that’s 92% of the entire genome) categories, with 38% and 28% fewer call errors than the second-best contestants, respectively. Fast forward to today, the DRAGEN team is introducing powerful machine learning (ML) and further improved graph genome mapping (expected to be available in early 2022, as a beta version in the DRAGEN v3.10 release). These new advancements propel DRAGEN to lead accuracy across all read technologies in all benchmark regions and the MHC region. In this blog, we show the results collected in the PrecisionFDA Truth Challenge V2, compare the latest DRAGEN accuracy against the challenge submissions across all read technologies and describe the methods used to reach high accuracy levels.
PrecisionFDA Truth Challenge v2
The PrecisionFDA Truth Challenge V2 aimed to assess the state-of-the-art of small variant calling on a common frame of reference, with a focus on benchmarking accuracy in difficult-to-map regions, segmental duplications, and the Major Histocompatibility Complex (MHC) region across reads produced by multiple sequencing platforms (~35X Illumina, ~35X PacBio HiFi, and ~50X Oxford Nanopore Technologies). The participants could use different technologies either individually or combined with a hybrid approach. As shown in Figure 1, the participants trained their methods on HG002 subject, using the HG002 FASTQs as input and benchmarking against the HG002 high confidence call set. Then they were blindly assessed using HG003 and HG004 datasets and associated high confidence call sets. Submissions were evaluated following best practices for benchmarking small variants with the new v.4.2.1 GIAB benchmark sets and genome stratifications.

Figure 1: PrecisionFDA Truth Challenge V2 overview1
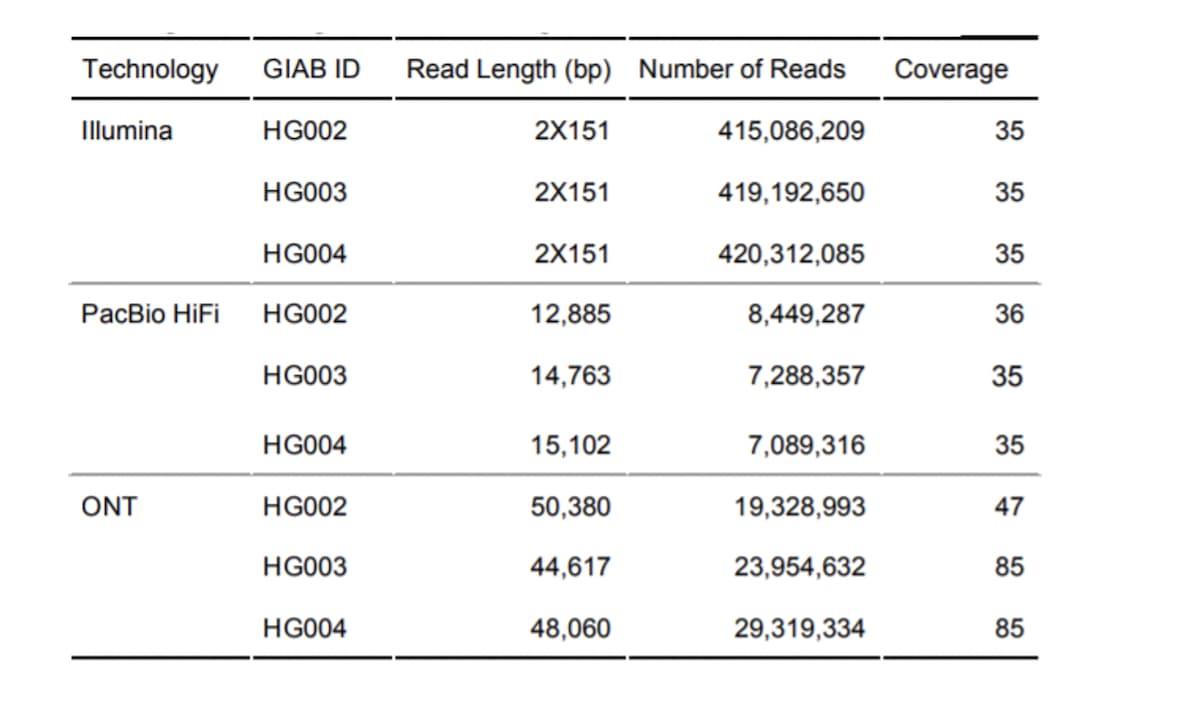
Table 1: Sequencing dataset characteristics, used in PrecisionFDA Truth Challenge V2. Read Length - N50 used to summarize PacBio and ONT read lengths. Coverage - median coverage across autosomal chromosomes.
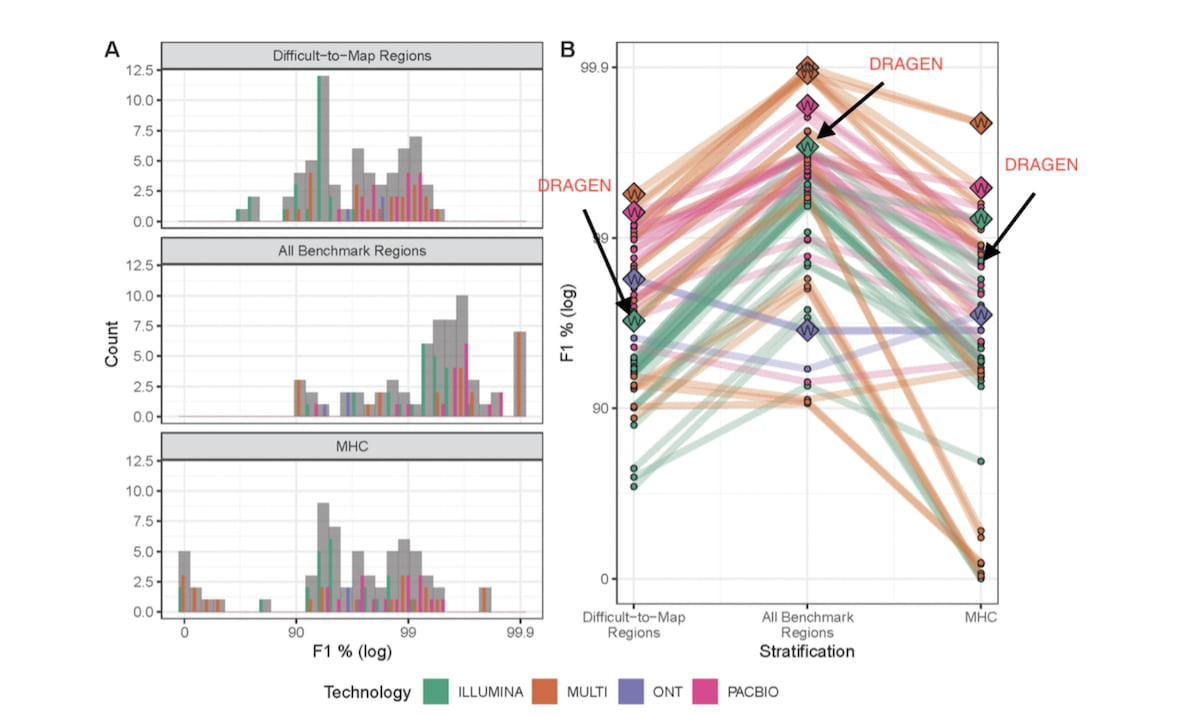
Figure 2: Results of the PrecisionFDA Challenge v2 – Summer 20202
Overall performance (A) and submission rank (B) varied by technology and stratification (log scale).
Generally, submissions that used multiple technologies (MULTI) outperformed single technology submissions for all three genomic context categories. Panel A shows a Histogram of F1 % (higher is better) for the three genomic stratifications evaluated. Submission counts across technologies are indicated by light grey bars and individual technologies by colored bars. Panel B shows individual submission performance. Data points represent submission performance for the three stratifications (difficult-to-map regions, all benchmark regions, MHC), and lines connect submissions. Category top performers are indicated by diamonds with “W”s and labeled with Team names.
DRAGEN 3.7 version competed in the Illumina reads category and ranked first in two of the three test regions (Difficult-to-Map Regions, and All-Benchmark Regions). DRAGEN 3.7, with graph enabled, significantly reduced false positives and false negatives compared to earlier versions of DRAGEN. Since then, we further innovated on DRAGEN and its methods, resulting in gains in accuracy that puts it ahead in some categories across all read technologies. We show below that the combination of further graph genome improvements and machine learning yields the highest accuracy as measured by the PrecisionFDA Truth challenge V2 in the All-Benchmark Regions and the MHC region.
DRAGEN Accuracy Improvements
The DRAGEN team has developed several key changes to improve variant calling accuracy over a larger portion of the human genome, while making sure that these improvements are generalizable to a wide population of samples. The first is improvements to the graph genome, the second is the development of alt-masking, and reference genome updates, and the third is improving the small variant caller using machine learning.
The DRAGEN graph genome contains population SNP’s and alternate haplotypes to enable more accurate read mapping. No PrecisionFDA truth subjects were included in the population that contributed variants to the DRAGEN graph genome. Recently, some improvements to the graph were made in the MHC regions, by covering a wider portion of the regions, with a greater diversity of population ALT haplotypes.
Portions of ALT contigs in a reference genome can be highly similar to portions of primary chromosome contigs, which can cause read mapping ambiguity and variant calling errors. This issue was addressed via ALT-masking: identifying such ALT regions and converting them to contiguous strings of “N” bases. The PrecisionFDA truth sets were not used in the main ALT-masking method which masked ~100Mb of ALT sequence. However, several clusters of ALT-masking-induced variant calling errors were observed in datasets from every NIST truth subject (HG001-7). The ALT-mask was adjusted to correct these errors.
The ML module first added in DRAGEN 3.9 and further improved in 3.10 employs a supervised model that uses contextual and read-based features extracted from the DRAGEN variant caller. Substantial gains were demonstrated consistently across all subjects, including test data from other populations that were not used during training.
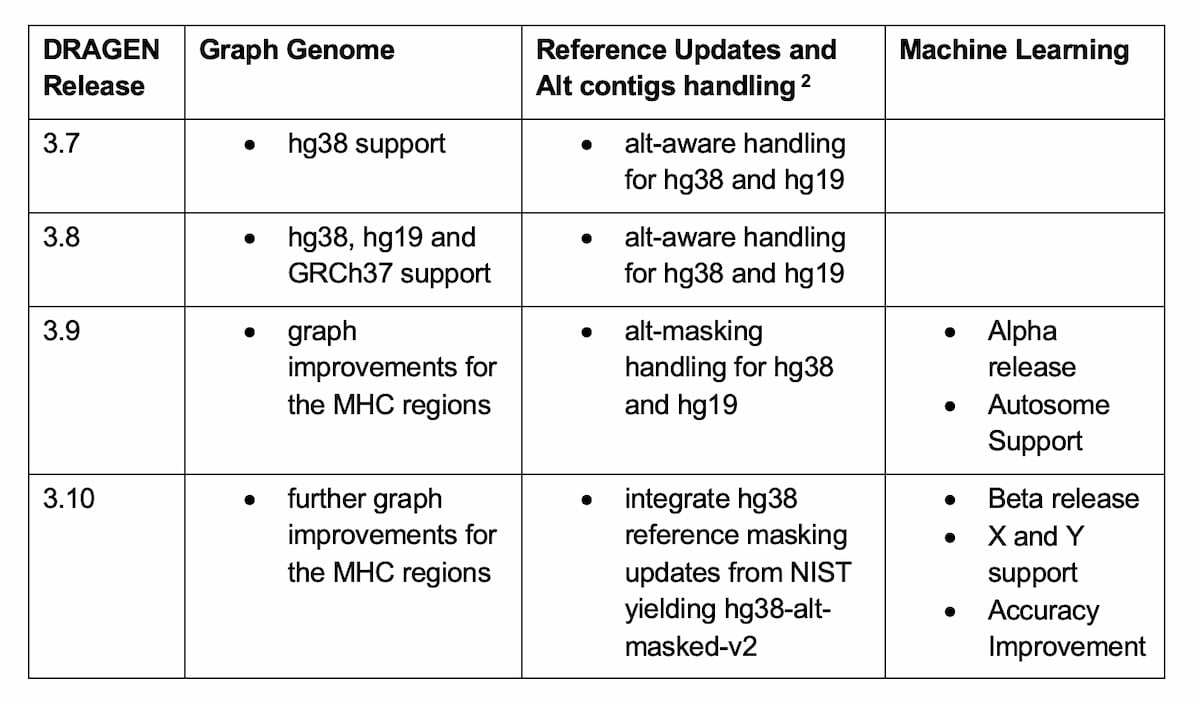
Table 2: Methods Improvements yielding better small VC accuracy in DRAGEN since the PrecisionFDA Truth Challenge v2.
DRAGEN 3.10 was run using the combination of these three methods updates on the HG003 and HG004 FASTQs from the PrecisionFDA Truth Challenge V2. The resulting VCFs were uploaded to the PrecisionFDA app to generate accuracy results according to the benchmarking guidelines. The resulting accuracy could be directly compared against the challenge submissions results. The comparison shows that DRAGEN 3.10+graph+ML outperforms all read technologies (Illumina, HiFi PacBio, and ONT) for the All-Benchmark Regions and the MHC regions.
Figure 3 shows the averaged HG003/HG004 F1 score for combined SNVs and INDELs for all submissions over the All-Benchmark regions. The original DRAGEN submission used DRAGEN 3.7+graph and came in 1st position at the time of the challenge for the Illumina reads category and is now at the 6th position overall. DRAGEN 3.10 + graph shows improvement over the DRAGEN 3.7+graph, thanks to graph and reference/alt-contig handling improvements, and DRAGEN 3.10+graph+ML comes in first place tied with the top performing HiFi PacBio read submission.
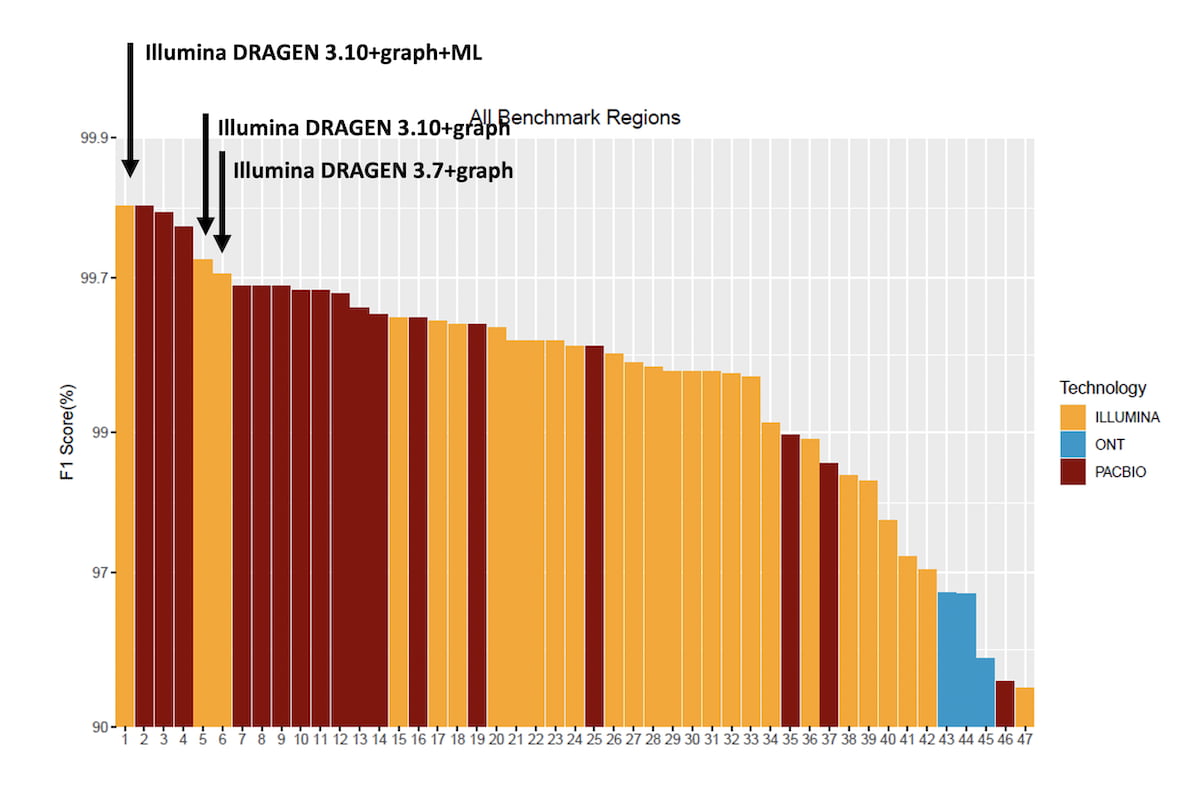
Figure 3: Accuracy of DRAGEN 3.10 Graph and ML compared to the PrecisionFDA Truth Challenge v2 submissions in the ‘All benchmark regions’.
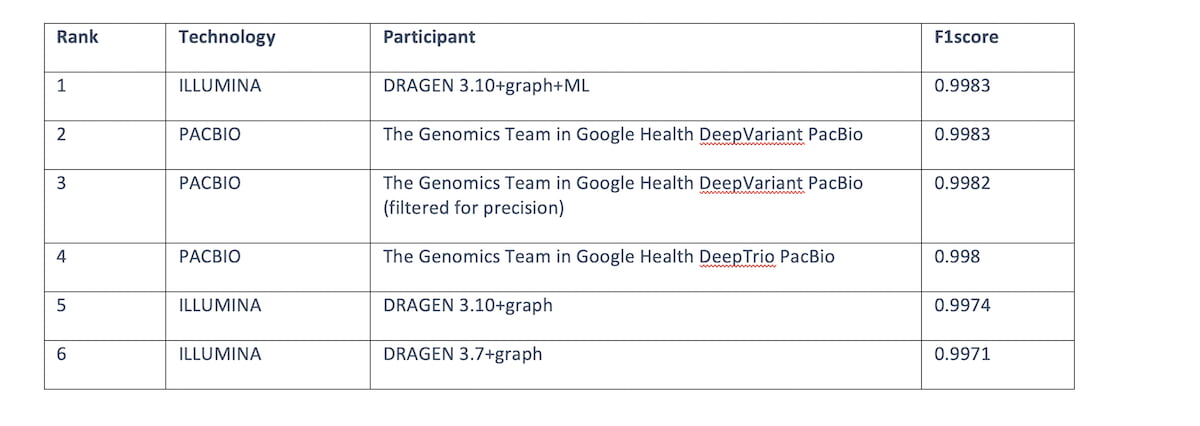
Table 3: Top 6 submissions F1 score across all read technologies for the “All benchmark regions”. DRAGEN 3.10+graph+ML comes in first place tied with HiFi PACBIO.
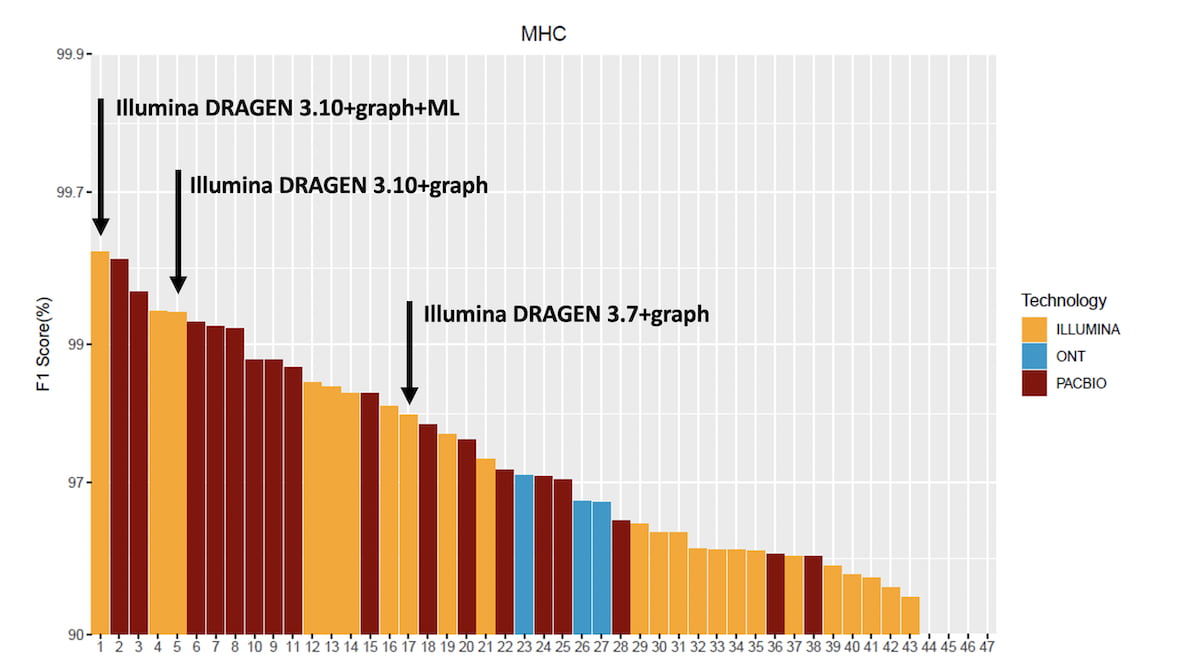
Figure 4: Accuracy of DRAGEN 3.10 Graph and ML compared to the precisionFDA v2 submissions in the MHC region.
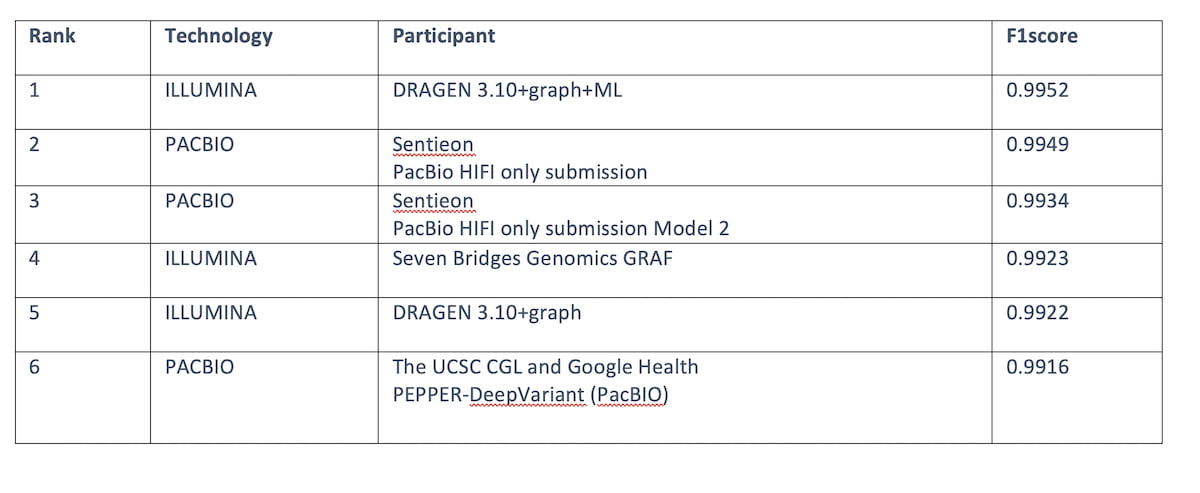
Table 4: Top 6 submissions F1 score across all read technologies for the “MHC regions”. DRAGEN 3.10+graph+ML comes in first place ahead of HiFi PacBio.
Machine Learning in DRAGEN
In DRAGEN v3.9, we added a powerful and efficient Machine Learning recalibration pipeline as an option within the germline small variant workflow. The pipeline runs the Machine Learning model after the standard variant calling when enabled. The ML step recalibrates the QUAL and GQ fields that are output to the final VCF. In some cases, ML can change GT. The pre-ML values of these fields are preserved in the DQUAL, DGT, and DGQ fields so that no information is lost.
The ML step only adds about five minutes for a 30x WGS germline run to the standard workflow, so the accuracy improvements have a limited impact on the total runtime.
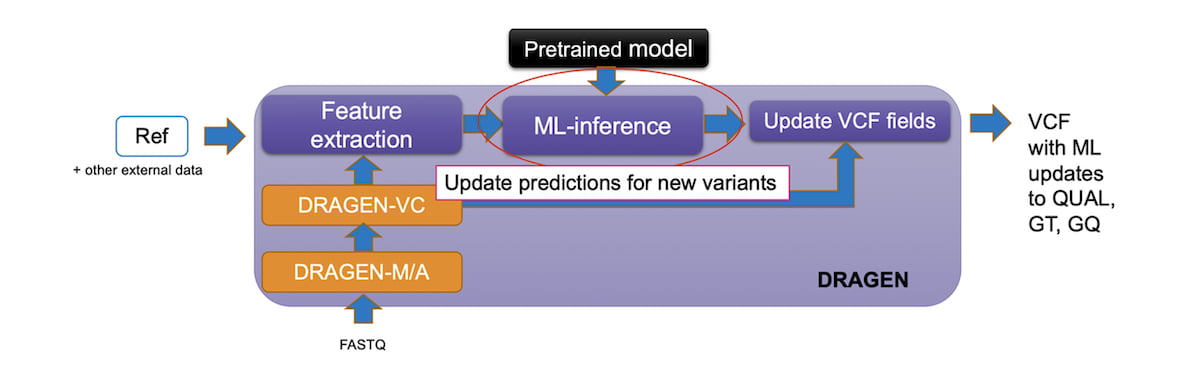
Figure 5: DRAGEN Germline Workflow with ML enabled.
The ML model is generated using supervised offline training. The model processes a set of read-based and contextual features to refine the accuracy of the small variant caller quality scores. The features used to train the model include Mappability, AF, VC-Qual, DP, GC content, mismatches and other internal mapping, alignment and VC metrics.
Figure 6 below shows the total number of SNP errors (FP+FN) collected over the v.4.2.1 “All benchmark” regions, for all 7 NIST subjects HG001-7. The results show two key findings: DRAGEN graph reduces the SNP errors by ~50% consistently across all 7 subjects, showing robustness across various ancestries (HG002-4 is a Ashkhenazi trio and HG005-7 is a Chinese trio). Further, DRAGEN ML yields an additional 20-30% SNP errors reduction, also consistently across all 7 subjects.
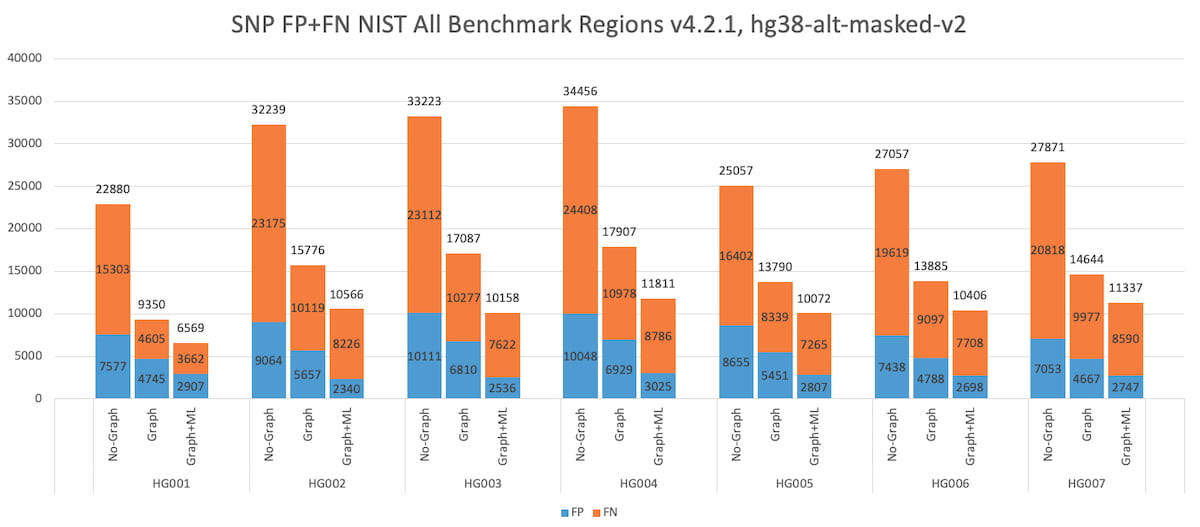
Figure 6: HG001 through HG007 SNP Accuracy Results in the extended truth set (v4.2.1 VCF and BED).
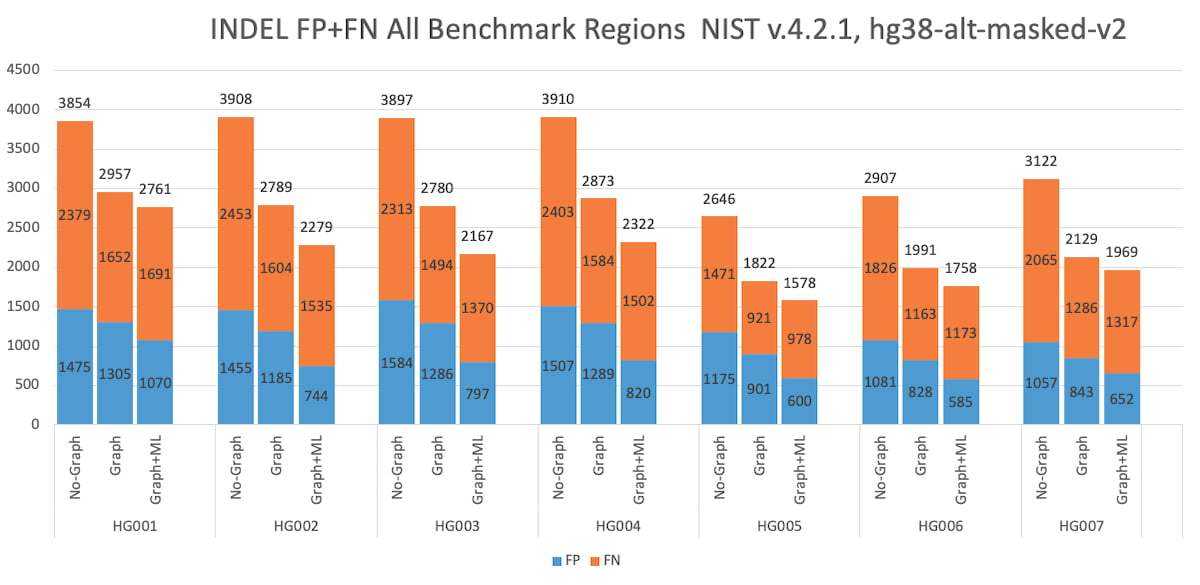
Figure 7: HG001 through HG007 INDEL Accuracy Results in the extended truth set (v4.2.1 VCF and BED).
Native Alt-Masking in DRAGEN
In DRAGEN 3.9, we introduced a new approach to handle native reference ALT contigs, where strategic positions of the ALT contigs are masked to increase accuracy. The ALT-Masking approach was introduced to replace the ALT-Aware alignment liftover procedure which improves accuracy.
While we achieved significant accuracy improvements in DRAGEN with liftover-based ALT awareness, testing over time unveiled stubborn issues. Mainly, long liftover alignments of 5Mbp sequences can sometimes be problematic. There are many places where the "correct" or most useful liftover between a long ALT haplotype and the primary assembly is ambiguous. Incorrect liftover can produce dense clusters of mismapped reads and false variant calls. From time to time, another place where bad liftover caused mapping and VC issues, were discovered, which tended to be local but severe. We introduced the ALT-masked-based solution to solve these issues.
Under the ALT-masked-based approach, segments similar to the primary assembly are masked, so they do not compete and steal alignments or squash MAPQs. Segments which are quite different are left unmasked, functioning essentially as decoy sequences. The PrecisionFDA truth sets were not used in the main ALT-masking method which masked ~100Mb of ALT sequence. However, several clusters of ALT-masking-induced variant calling errors were observed in datasets from every NIST truth subject (HG001-7). The ALT-mask was adjusted to correct these errors. DRAGEN’s ALT masked references deliver improved variant calling accuracy vs. the liftover-based ALT-aware method. The base masking approach has the benefits of using ALT contigs without the negative consequences. It is also easier to define, maintain, and improve. We will likely continue to refine the masks over time, but they've already surpassed liftover-based performance. More details can be found in the article on the reference improvements in DRAGEN3.
Improvements in the MHC region
DRAGEN has gained a considerable boost in small variant calling accuracy in the MHC region since the 3.7 version, via the combination of ML, ALT-masking, and graph-reference improvements. The graph-based reference improves the mapping and variant calling accuracy in difficult-to-map regions of the genome, such as the MHC region. The MHC region is highly polymorphic, and sample reads differ greatly from the reference making it difficult for the mapper to find a good match. Mapping can be improved by adding carefully chosen population haplotype segments that usefully distinguish among homologous regions and provide alternate paths known to the population to the linear reference.
DRAGEN has improved the graph reference in the MHC region by enriching the population haplotypes that cover the MHC region. This leads to improved mapping accuracy and helps boost the variant calling accuracy, and the addition of ML enables DRAGEN to have a higher F1 score than the PrecisionFDA Challenge v2 submissions. The graph reference hash-table can be downloaded from the Illumina DRAGEN support page4.
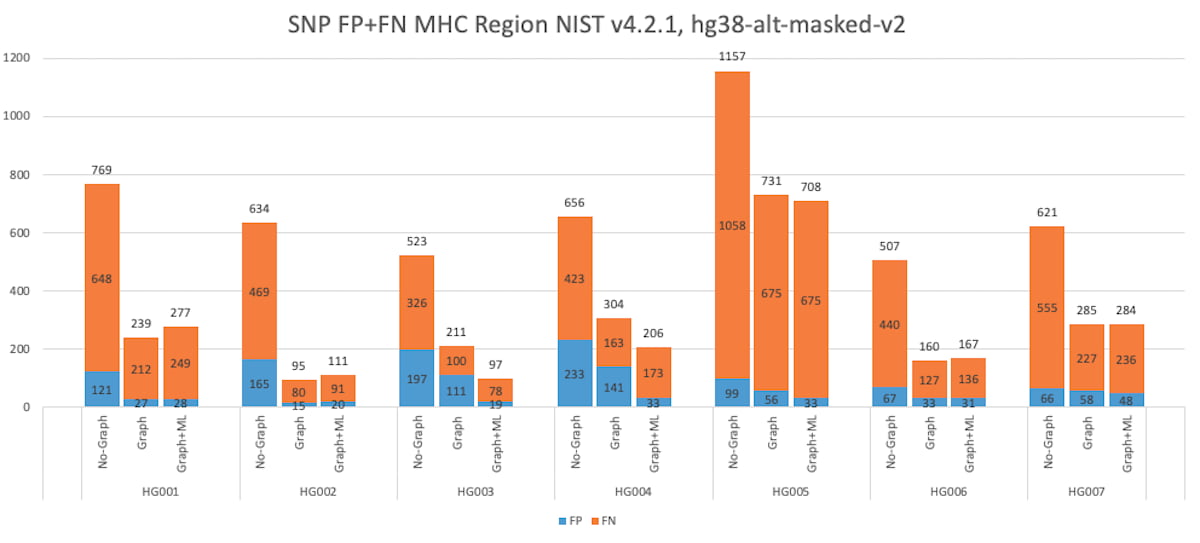
Figure 8: HG001 through HG007 SNP Accuracy Results in the MHC truthset (v4.2.1 VCF and BED).
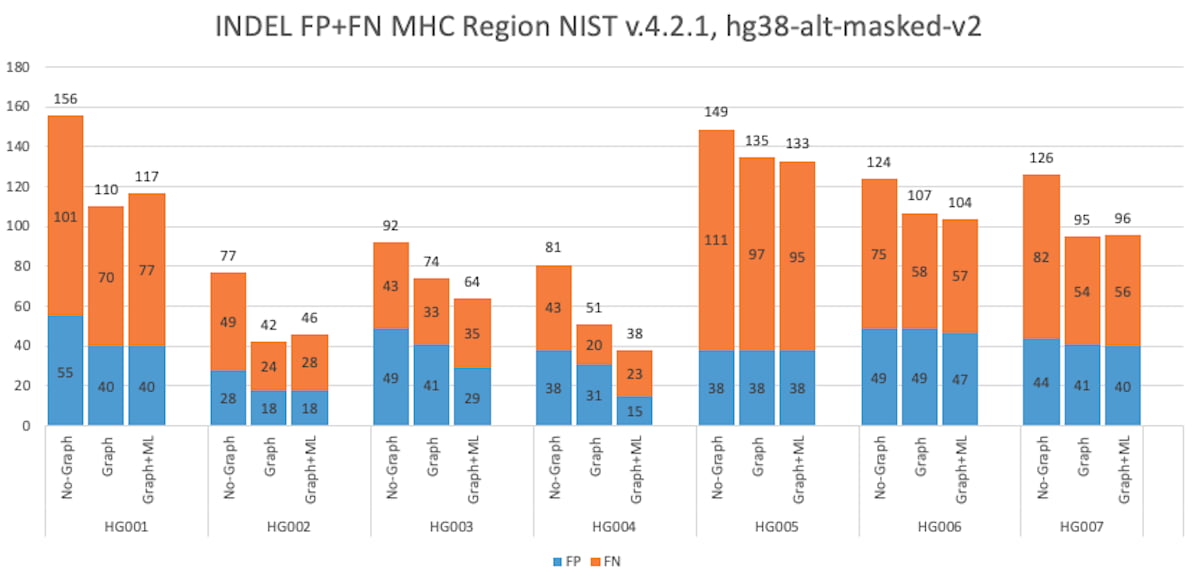
Figure 9: HG001 through HG007 INDEL Accuracy Results in the MHC truthset (v4.2.1 VCF and BED).
DRAGEN: Driving Continuous Innovation & Advancing Genomic Analysis
DRAGEN is a platform that provides highly accurate, comprehensive, and ultra-rapid secondary analysis at scale, for NGS data processing. The continuous accuracy improvements, and an expanded coverage into the difficult regions of the genome are critical assets for a comprehensive genomic solution, enabling the detection of challenging and medically relevant variants.
This article shows that improvements in the upcoming DRAGEN 3.10 release yield competitive small variant accuracy across all read technologies. Coupled with the rest of the DRAGEN variant callers’ suite (SV, CNV, Expansion Hunter, and targeted callers such as SMN, CY2D6 and HLA), DRAGEN enables the coverage of the entire genome, advancing genomic analysis.
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
References
- https://precision.fda.gov/challenges/10
- Olson et al. PrecisionFDA Truth Challenge V2: Calling variants from short- and long-reads in difficult-to-map regions BioRxiv. (2020) doi:2020:11.13
- https://www.illumina.com/science/genomics-research/articles/dragen-demystifying-reference-genomes.html
- https://support.illumina.com/sequencing/sequencing_software/dragen-bio-it-platform/product_files.html