Original description
Guillaume Duchenne de Boulogne should be remembered as one of the most influential medical scientists of the 19th century, who made pioneering contributions to neurology throughout his career. He was among the first to perform muscle biopsies and use photography for clinical purposes, and he invented devices to perform nerve conduction tests.
Among the patients in his care was a series of young boys suffering from a particularly devastating loss of muscle, which started with difficulty walking and eventually led to death due to the failure of diaphragm musculature, which prevents breathing. This condition was not widely recognized until Duchenne published a book including their stories in 1861. The condition now bears his name: Duchenne muscular dystrophy.
Clinical signs and natural history of DMD-related conditions
DMD is one of the largest known human genes, and is located on the X chromosome at cytobands Xp21.2-p21.1. It encodes dystrophin, part of the large dystrophin-associated glycoprotein complex that plays a critical role in maintaining the link between the cytoskeleton and the extracellular matrix, and thereby in preserving the integrity of the muscle cell membrane.1
There are three different conditions associated with DMD:
- People with little or no DMD gene activity have Duchenne muscular dystrophy (DMD). DMD is a severe, progressive, muscle-wasting disease. The earliest symptoms are difficulty climbing stairs, a waddling gait, and frequent falls; patients present with these symptoms in early childhood. Most patients become wheelchair dependent around 10–12 years of age and need assisted ventilation around age 20. Life expectancy for people with DMD is between 20 and 40 years.2
- People with partial DMD gene activity may have Becker muscular dystrophy (BMD). BMD is characterized by later-onset skeletal muscle weakness. Onset varies widely from five to 60 years of age; the course is slower and less predictable than that of DMD and the life expectancy is mid-forties.3
- Mothers who are carriers of DMD pathogenic variants are at risk of developing DMD-associated dilated cardiomyopathy (DCM). This condition is characterized by a hypokinetic, dilated phenotype and congestive heart failure progressively increasing with age.
The severe DMD phenotype is usually caused by variants disrupting the reading frame leading to the complete loss of dystrophin expression, whereas the less severe BMD phenotype is typically associated with in-frame mutations leading to the production of abnormal yet semi-functional protein (with intact N and C termini).4
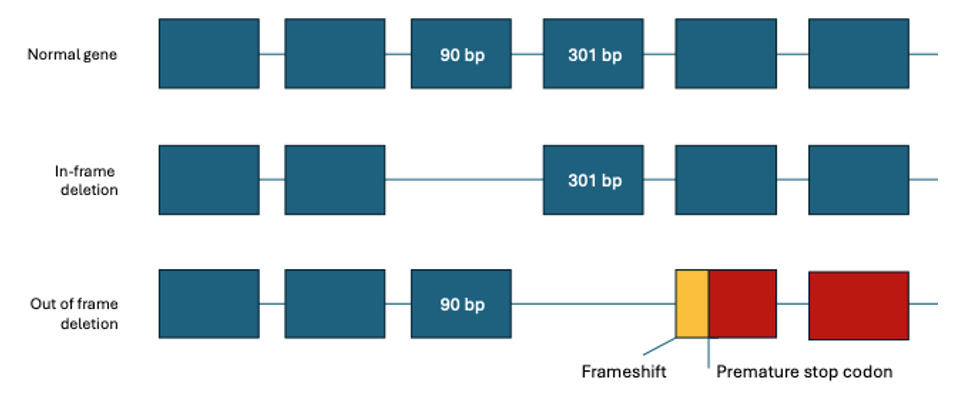
Mutation analysis has traditionally been challenging, due to the large gene size (79 exons over 2.2 Mb of genomic DNA) and the diversity of mutation types. Most identified mutations are deletions, accounting for 60%–65% of DMD and 85% of BMD mutations, and duplications have been observed in approximately 5%–10% mutations. The remaining causes are small mutations such as indel, point mutation, or splicing mutation.6 It has been estimated that about 25%–33% of all cases result from de novo mutational events.7
What causes Duchenne muscular dystrophy?
Because nearly all cases of DMD are boys and multiple people can be affected within the same family but only if they shared a maternal relative, it was originally presumed that DMD must be caused by a variant on the X chromosome. This proved to be true, and the gene was cloned successfully by Lou Kunkel and collaborators in 1987.8 This was among the vanguard of early disease gene discoveries, falling in between HBB for sickle cell anemia (1985) and CFTR for cystic fibrosis (1989).
While single-nucleotide variants and small insertions or deletions under 50 bp can certainly cause DMD, 80% of pathogenic variants in DMD are deletions or duplications of one exon or greater.9
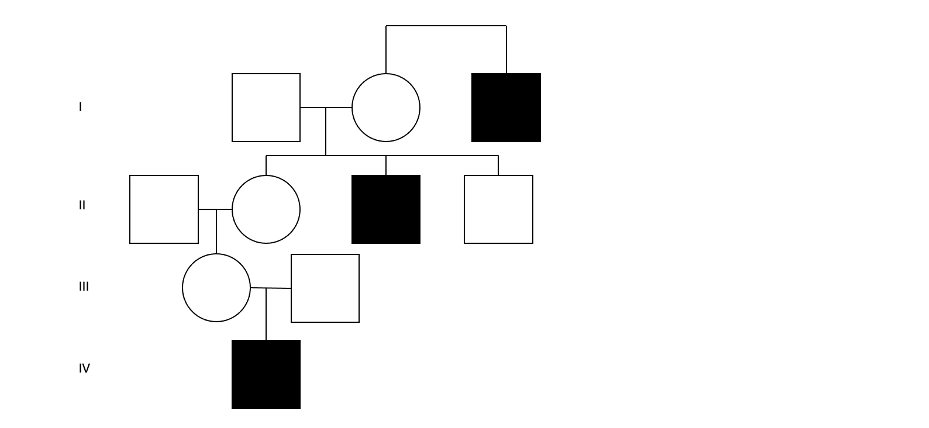
Figure 2: A hypothetical four-generation pedigree with three individuals affected by DMD—This demonstrates the typical pattern of X-linked recessive inheritance: multiple generations of affected males sharing a maternal lineage.
DMD therapies
Classic symptomatic interventions, such as braces and corticosteroids, are beginning to give way to precision medicine approaches. For affected individuals with specific exon deletion events, antisense oligonucleotide drugs can be used to induce skipping of specific remaining exons to restore partial DMD gene function.10 This approach is highly dependent on having an accurate molecular diagnosis.
In 2023, a more general gene therapy was approved for DMD: Elevidys. This product uses a viral vector to introduce a shortened version of the DMD gene into recipients, causing their own muscle cells to produce “micro-dystrophin.” This compact form of the gene product contains all necessary structural and functional components, and the Food and Drug Administration has concluded that the data from its clinical trial demonstrates that this effect “is reasonably likely to predict clinical benefit” in young patients. This gives hope that gene therapy approaches to this and other conditions are here to stay.
How are DMD variants typically detected?
Multiplex ligation dependent probe amplification (MLPA) is a popular technique for analyzing copy number variants (CNV) that has been widely applied for DMD. This amplification-based approach is effective at detecting the most common pathogenic DMD variants, but has several key drawbacks, including the inability to detect single nucleotide variants (SNV) and small indels.
Recent advancements in next-generation sequencing (NGS) and improvements in bioinformatics algorithms ultimately resulted in the development of a single NGS-based assay to detect all variant types, including CNV and sequence variants. Full-sequence analysis of the entire gene allows the detection of deep intronic pathogenic variants and accurate breakpoint detection of CNVs involving similar exons, which could impact the outcome of clinical trials.11
While standard approaches as described can detect most of the variants affecting coding regions, sub-exonic, sub-intronic, deep intronic, or large structural arrangements like inversions may go undetected, with 2%–7% of DMD cases remaining unresolved.12 Clinical whole-genome sequencing, however, can capture most genetic variants across both protein-coding and non-protein-coding regions, including SNVs, CNVs, and small structural variants (SVs).
How does the Genome Era change the game for DMD?
Whole-genome sequencing (WGS) offers the promise to resolve these limitations through a combined approach assessing changes in coverage depth with analysis of reads that span breakpoints. Because WGS is consistent from lab to lab and subject to subject—as opposed to hybrid capture or amplicon-based sequencing, which can vary greatly across both axes—methods developed to detect these genomic rearrangement events can be utilized by anyone. See our previous blog post about joint CNV-SV calling for more information about these methods.
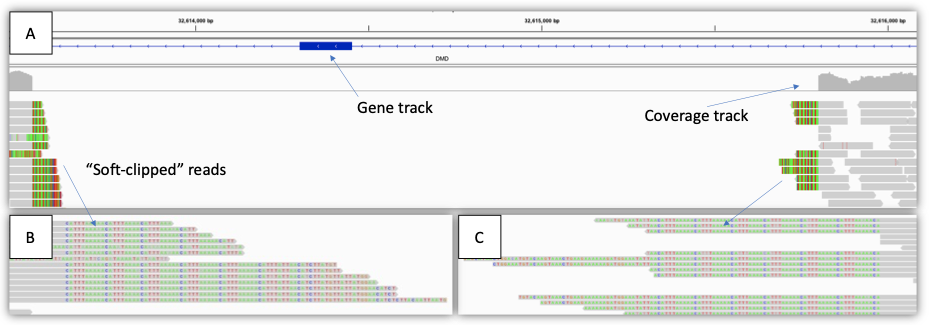
Figure 3: Example of a single exon deletion (2 kb) in a male with Duchenne muscular dystrophy—(A) IGV view showing the region impacted, including gene track, coverage track, and aligned reads track. Note that the coverage drops to zero across the region, indicating a hemizygous deletion. (B),(C) zoomed-in IGV view showing the “soft-clipped” reads. The gray segments of each read match the reference sequence, whereas the colored letters do not (soft-clipped). In this case, the soft-clipped reads map to the other end of the deletion breakpoint. These reads can be used to generate SV calls.
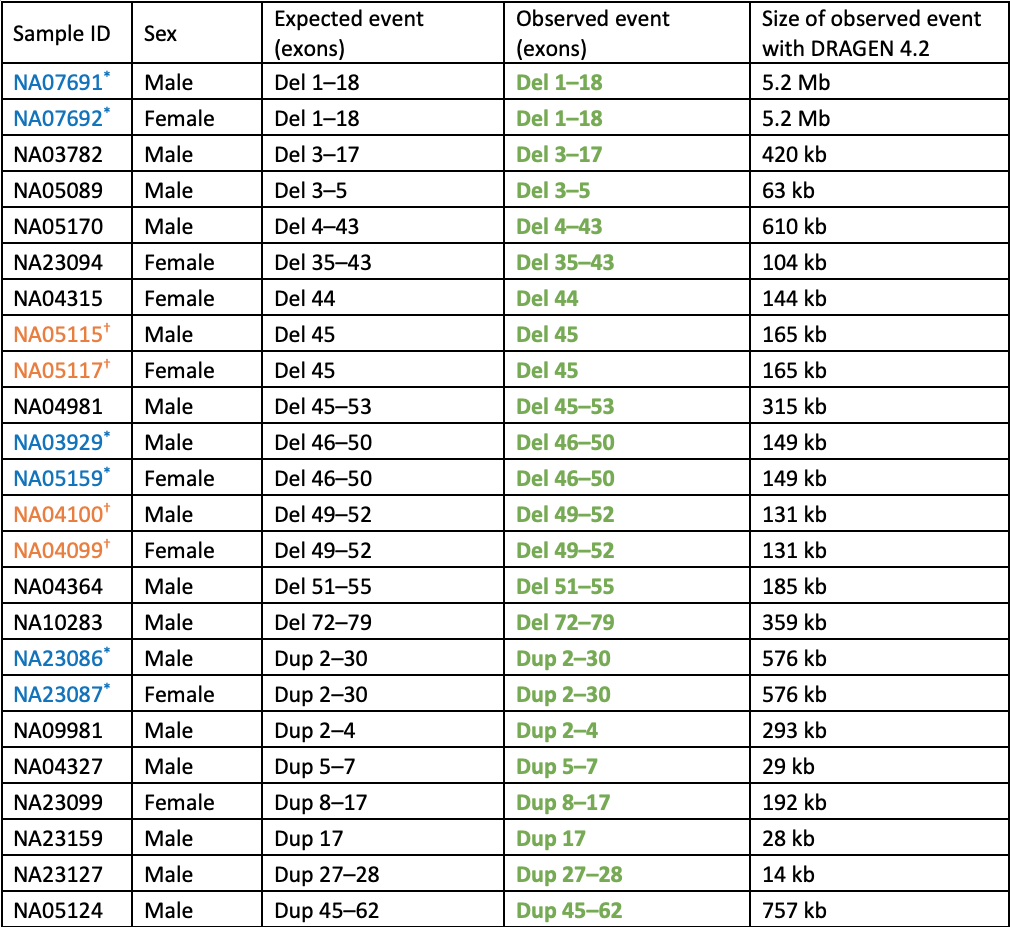
Table 1: Cell lines with known DMD copy number variants analyzed by WGS and DRAGEN 4.2 Joint SV caller—All expected events were detected. Consecutive entries where the sample ID is set in blue or orange with an * or † are duos where the male is the son of the female carrier. Bolded and set in green are events matching the expected exons, CNV type (del or dup), and zygosity. All events in females are heterozygous and in males are hemizygous.
Notably, no false positive exonic DMD copy number events were called for any of the remaining ~3200 individuals in the 1000 Genomes Project dataset. This demonstrates a very low false positive rate for this variant type overall, which is key for high-throughput projects such as population genomics or carrier screening research.
Conclusions
Joint CNV and SV calling using WGS and DRAGEN is an extremely sensitive combination, capable of detecting medically relevant deletions and duplications in DMD. As this gene is perhaps the most challenging gene known for CNV calling, especially in female carriers, this evidence shows that WGS and DRAGEN 4.2 set a very high bar. These outputs are compatible as input to the Emedgene tertiary analysis and research reporting software, which includes explainable AI-powered variant prioritization for small genomic variants such as SNVs and indels, mitochondrial DNA variants, and even short tandem repeats. This means that you can use Illumina tools from sample to answer for seamless research evaluation of DMD.
And because this is a generalized approach, other genes with medically relevant exon-level CNVs can also be evaluated using these methods. Some examples where similar variants are crucial for testing include LDLR for familial hypercholesterolemia, NF1 for neurofibromatosis, BRCA1/2 for hereditary breast and ovarian cancer, and PRKN for early-onset Parkinson disease.
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
References
- Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem. 1990 Nov;108(5):748-52. doi:10.1093/oxfordjournals.jbchem.a123276
- Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-2038. doi:10.1016/S0140-6736(19)32910-1
- Darras BT, Urion DK, Ghosh PS, et al. Dystrophinopathies. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews. 2000 Sep 5. PMID:20301298
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1998 Jan;2(1):90-5. doi:10.1016/0888-7543(88)90113-9
- Le Rumeur E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn J Basic Med Sci. 2015 Jul 20;15(3):14-20. doi:10.17305/bjbms.2015.636
- Ji X, Zhang J, Xu Y, et al. MLPA Application in Clinical Diagnosis of DMD/BMD in Shanghai. J Clin Lab Anal. 2015;29(5):405-411. doi:10.1002/jcla.21787
- Santos R, Gonçalves A, Oliveira J, et al. New variants, challenges and pitfalls in DMD genotyping: implications in diagnosis, prognosis and therapy. J Hum Genet. 2014;59(8):454-464. doi:10.1038/jhg.2014.54
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509-517. doi:10.1016/0092-8674(87)90504-6
- Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395-402. doi:10.1002/humu.22758
- Matsuo M. Antisense Oligonucleotide-Mediated Exon-skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021;4(3):232-240. doi:10.31662/jmaj.2021-0019
- Nallamilli BRR, Chaubey A, Valencia CA, et al. A single NGS-based assay covering the entire genomic sequence of the DMD gene facilitates diagnostic and newborn screening confirmatory testing. Hum Mutat. 2021;42(5):626-638. doi:10.1002/humu.24191
- Andrews JG, Galindo MK, Thomas S, Mathews KD, Whitehead N. DMD Gene and Dystrophinopathy Phenotypes Associated With Mutations: A Systematic Review for Clinicians. J Clin Neuromuscul Dis. 2023;24(4):171-187. doi:10.1097/CND.0000000000000436