Key takeaways
An internal analysis evaluated the NovaSeq X Series against the Ultima Genomics UG 100 platform. This evaluation found that the NovaSeq X Series:
- Measures accuracy of WGS against the full NIST v4.2.1 benchmark, while Ultima Genomics measures WGS accuracy using the UG 100 platform against a subset that excludes 4.2% of the genome
- Results in 6× fewer SNV errors and 22× fewer indel errors than the UG 100 platform, when assessed against the full NIST v4.2.1 benchmark
- Calls approximately 180,000 more SNVs and 270,000 more indels analyzing the whole genome compared to the UG 100 platform analyzing only the Ultima Genomics "high confidence region"
- Maintains high coverage and variant calling accuracy in repetitive genomic regions, including GC-rich sequences and homopolymers longer than 10 base pairs, compared to the UG 100 platform
- Provides more insights into biologically relevant genes compared to the UG 100 platform
Ultima Genomics vs. Illumina sequencing platforms
Illumina has maintained a relentless commitment to innovating next-generation sequencing (NGS) capabilities and building future methods.1 The NovaSeq X Series enables data-intensive applications at production scale to empower scientists to make new discoveries. Over the past decade, emerging companies like Ultima Genomics have introduced NGS platforms. In 2024, Ultima Genomics launched the UG 100 sequencing platform with bold claims, including that it is capable of sequencing 20,000 human genomes per year with industry-leading accuracy in variant calling.2 To evaluate these performance and accuracy claims, Illumina conducted a comparative analysis of the NovaSeq X Series and the UG 100 platform. The results of this analysis demonstrate that the NovaSeq X Series delivers more accurate variant calling, provides a higher-quality, more comprehensive genome, and enables more insights into the molecular mechanisms of disease than the UG 100 platform.
Data sources for comparing Ultima Genomics to Illumina
We generated Illumina whole-genome sequencing (WGS) data on the NovaSeq X Plus System using the NovaSeq X Series 10B Reagent Kit, followed by DRAGEN v4.3 secondary analysis. Data was downsampled to 35× coverage depth (including duplicates). We sourced Ultima Genomics WGS data from a publicly available data set released in a variant call format (VCF) that had been generated on the UG 100 platform at 40× coverage depth excluding duplicates, and analyzed using DeepVariant software by Ultima Genomics.
Higher variant calling accuracy with the NovaSeq X Series
The National Institute of Standards and Technology (NIST) v4.2.1 benchmark for the Genome in a Bottle (GIAB) HG002 reference genome is used to assess the accuracy and performance of WGS and variant calling analysis tools.3,4 The NIST v4.2.1 benchmark includes high-confidence genotype calls for single-nucleotide variants (SNVs), insertions and deletions (indels), and structural variants (SVs), along with challenging regions consisting of segmental duplications, low-mappability regions, or repetitive sequences that can include genes of biological interest.4,5
Illumina measures the accuracy of WGS obtained with the NovaSeq X Series with DRAGEN secondary analysis against the full NIST v4.2.1 benchmark, including all genomic regions (Figure 1).6 Conversely, Ultima Genomics measures the accuracy of WGS with the UG 100 platform against a defined subset of the NIST v4.2.1 benchmark referred to as the UG “high-confidence region” (HCR). The UG HCR masks regions with low performance on the UG 100 platform, including homopolymers, repetitive sequences, and areas with low coverage (Figure 1).7 Importantly, the regions excluded from the UG HCR include 4.2% of NIST benchmark variants.

Figure 1: Ultima Genomics HCR masks 4.2% of the genome─WGS accuracy on the NovaSeq X Series is measured against the full NIST v4.2.1 benchmark, while the UG HCR excludes 4.2% of the genome from analysis.
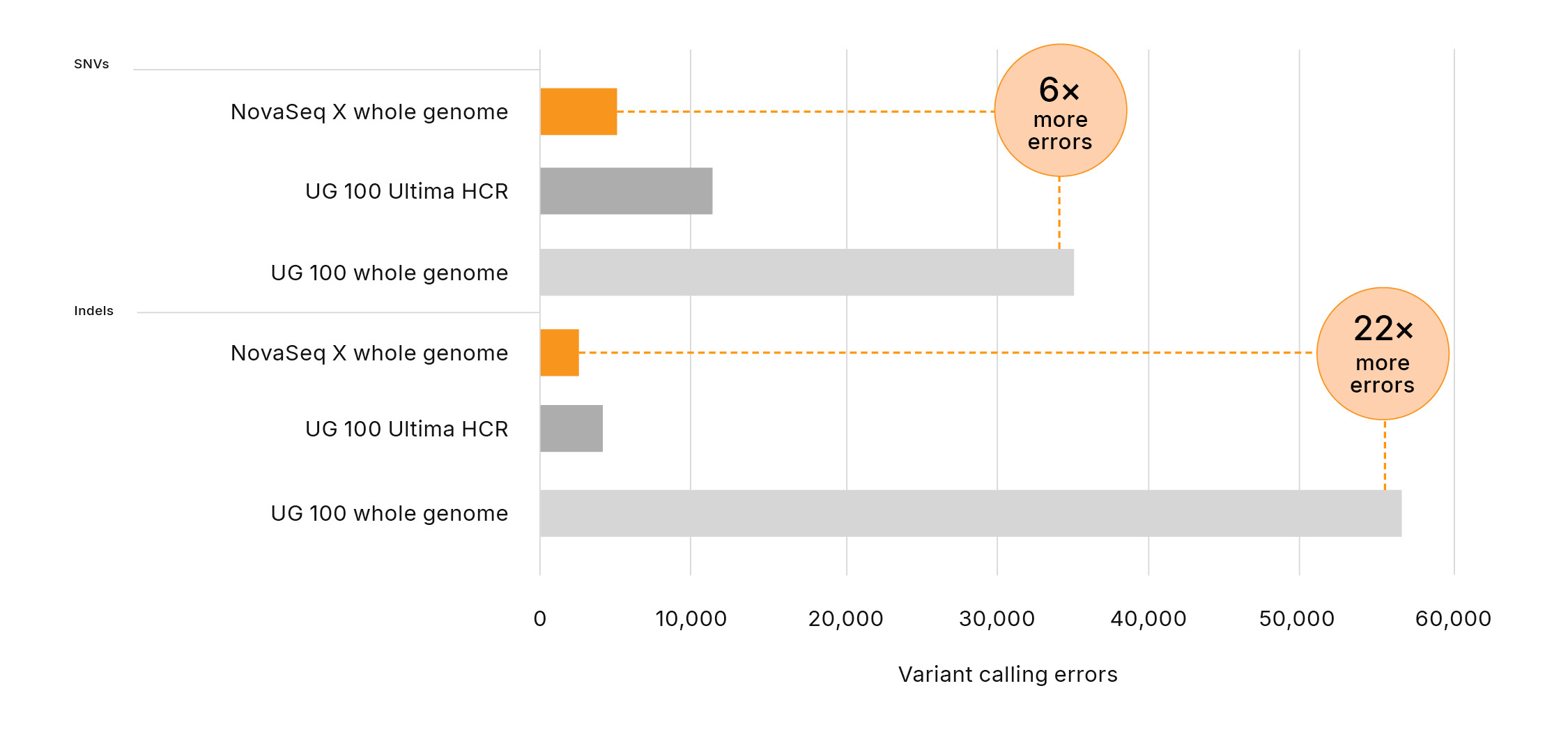
Figure 2: Significantly more errors in variant calling with Ultima Genomics─The UG 100 platform produces 6× and 22× more errors in variant calling for SNVs and indels, respectively, compared to the NovaSeq X Series, assessed against NIST v4.2.1 all benchmark regions. Errors are defined as the combined number of false positives and false negatives.

Figure 3: Ultima Genomics HCR masks key variants─Limiting analysis of WGS on the UG 100 platform to the UG HCR results in fewer variant calls for SNVs and indels compared to the NovaSeq X Series.
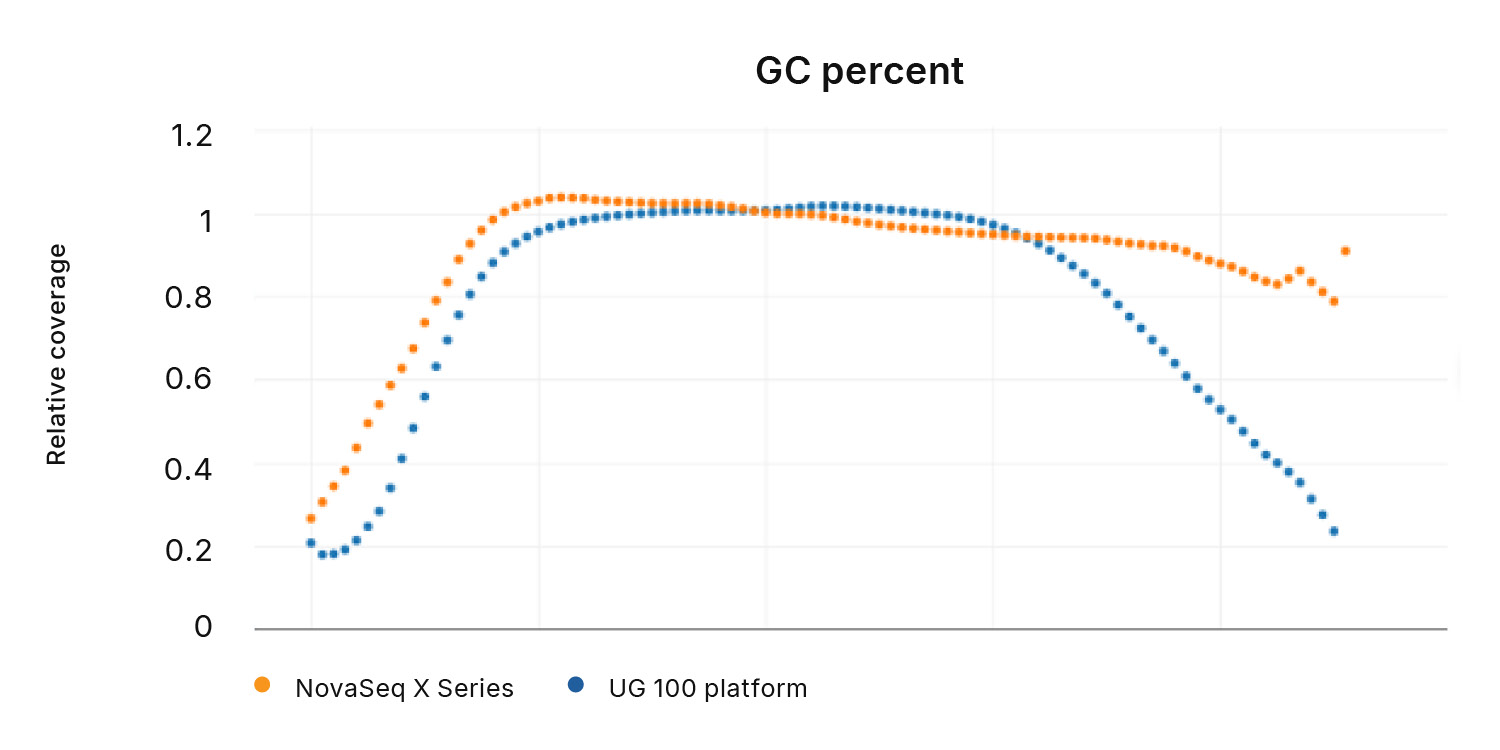
Figure 4: Loss of coverage in GC-rich regions with Ultima Genomics─The UG 100 platform loses coverage in repetitive, GC-rich regions of the genome compared to the NovaSeq X Series.
Increased genome coverage with the NovaSeq X Series
Illumina sequencing on the NovaSeq X Series delivers a comprehensive, high-quality genome that accurately calls 99.94% of SNVs,* 97% of copy number variants (CNVs),† 88% of SVs,ǂ 95.2% of short tandem repeats (STRs),§ at least 13 targeted gene callers for medically relevant genes with 98% concordance,¶ and human leukocyte antigen (HLA) typing with 94% concordance.**
* Compared against HG002 NIST v4.2.1 with rtg vcfeval.
† Compared against HG002 NIST v0.6 (hg38) with wittyer v0.4.1.
ǂ Compared against HG002 NIST T2T Q100 v1.1_v0.019 (hg38) with Truvari v.4.2.2.
§ 28 STR loci included in evaluation across 359 samples (157 samples with expansions in at least one locus).
¶ Concordance metrics against orthogonal technology sourced from published technical papers.
** Compared against T1K over 3202 diverse WGS samples from The 1000 Genomes Project.
In contrast, Ultima Genomics masks performance deficits by assessing the accuracy of WGS with the UG 100 relying on the UG HCR, which excludes regions of the NIST v4.2.1 benchmark that exhibit unreliable performance and confidence using Ultima Genomics sequencing technology. The extensive number of variants (about 450,000) not called by limiting analysis to the UG HCR (Figure 3) underscores the uncertainty about what data could be missed.
The regions excluded by the UG HCR amount to 4.2% of the genome, including 2.3% of the exome, and 1.0% of ClinVar variants. These excluded regions also account for 5.1% of genomic CNVs and 4.7% of ClinVar CNVs. Together, pathogenic variants in 793 genes are excluded from the UG HCR, limiting insight into the associated diseases. Furthermore, while Ultima Genomics claims high variant calling accuracy for SNVs and indels while sequencing through homopolymers, internal analysis showed that indel accuracy with the UG 100 platform decreased significantly with homopolymers longer than 10 base pairs, compared to the NovaSeq X Series (Figure 5). Indeed, the UG HCR excludes homopolymer regions longer than 12 base pairs.7 Given that published studies show that homopolymer repeat length may modulate the expression of nearby genes by altering nucleosome positioning,8 excluding these regions from Ultima Genomics analysis could result in valuable, biologically relevant insights being missed.
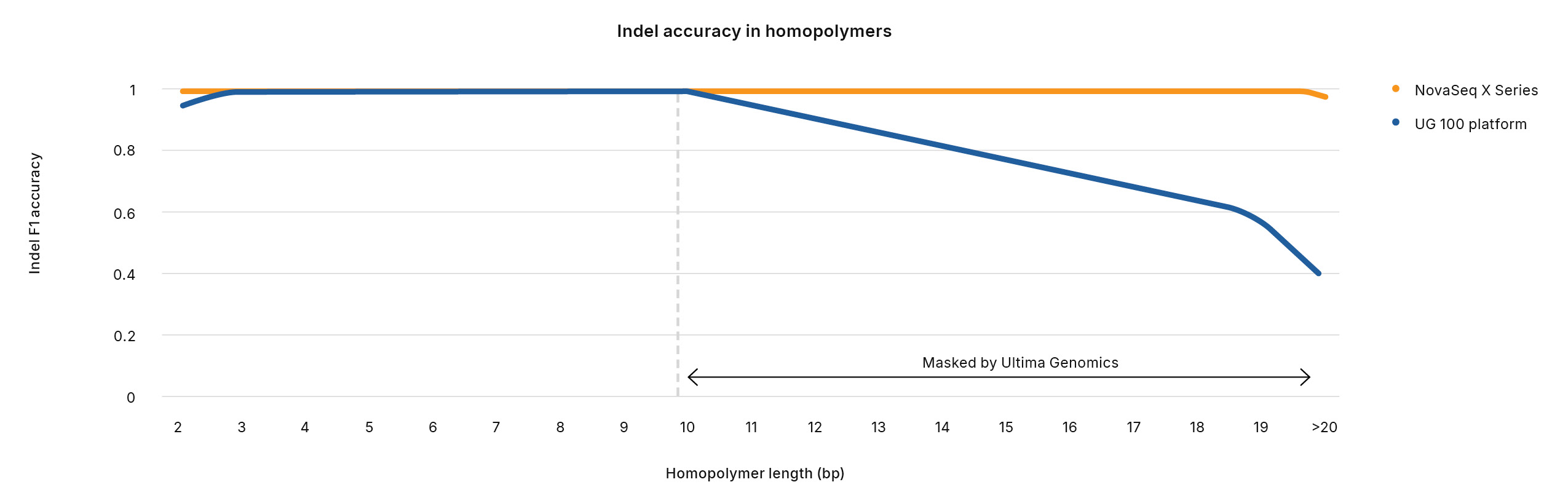
Figure 5: Reduced indel accuracy with Ultima Genomics─Indel accuracy with the UG 100 platform drops significantly with homopolymers longer than 10 base pairs, compared to the NovaSeq X Series.
More biologically relevant insights with the NovaSeq X Series
Genomic regions excluded from the UG HCR include functionally important loci in disease-related genes and STR-rich genes tied to immune and neurological traits (Table 1). One example is the B3GALT6 gene, which encodes an enzyme crucial to synthesis of glycosaminoglycans (GAGs). Variants in B3GALT6 have been linked to Ehlers-Danlos syndrome.9 Another example is the fragile X messenger ribonucleoprotein 1 (FMR1) gene, which is crucial to normal brain development and reproductive function. Alterations in FMR1 cause fragile X syndrome.10 Both B3GALT6 and FMR1 have GC-rich sequences that resulted in loss of coverage with the UG 100 platform, which likely excluded pathogenic and likely pathogenic variants in these genes (Figure 6A, 6B). Additionally, variants in the BRCA1 tumor suppressor gene have a well-established link to predisposing an individual toward breast cancer development.11,12 However, 1.2% of pathogenic BRCA1 variants are excluded from the UG HCR, and sequencing with the UG 100 platform resulted in significantly more indel calling errors in the BRCA1 gene compared to the NovaSeq X Series (Figure 6C).
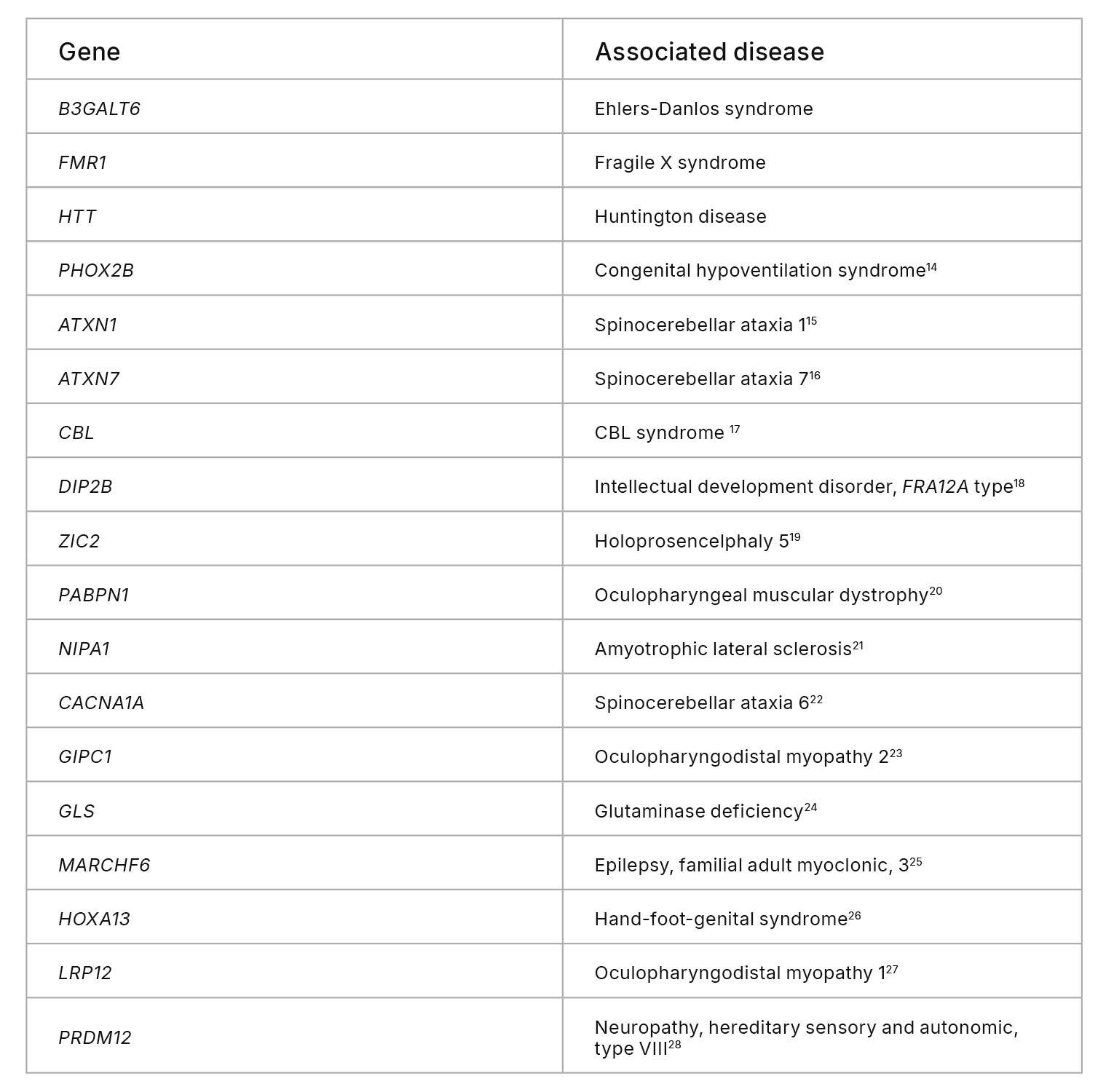
Table 1: Biologically relevant genes excluded from the UG HCR

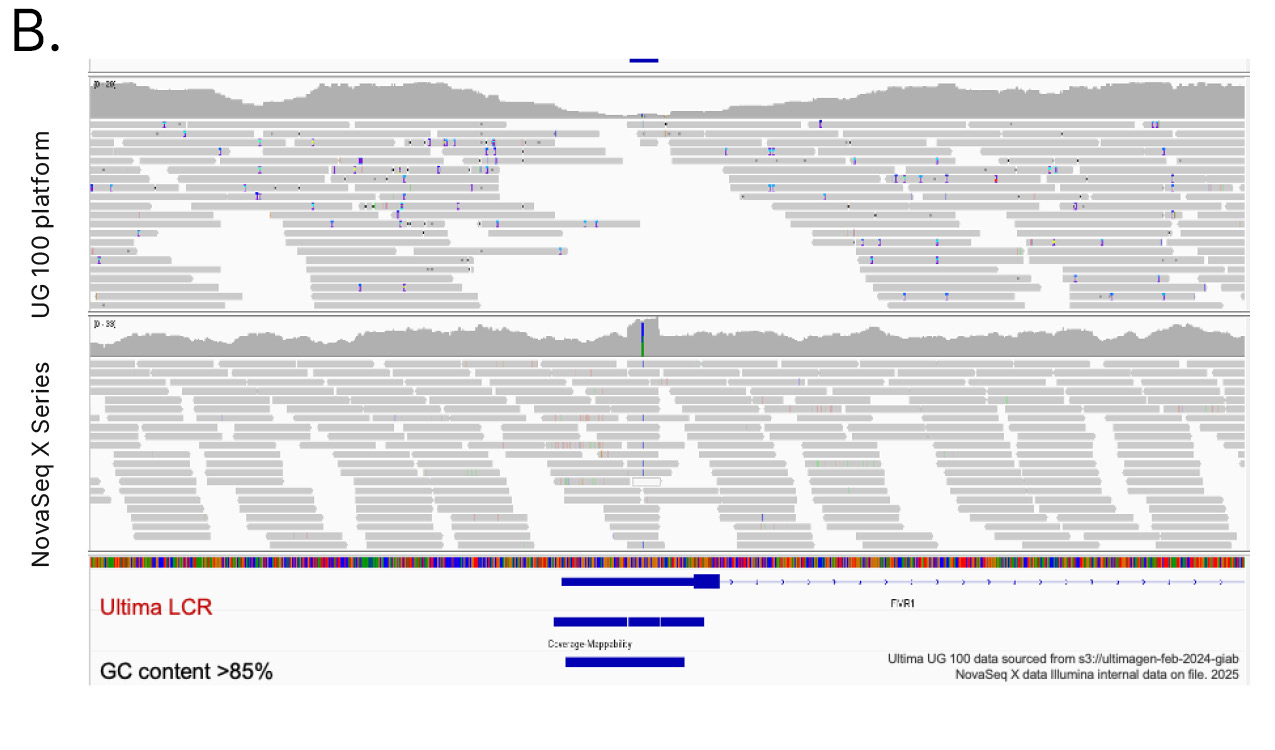
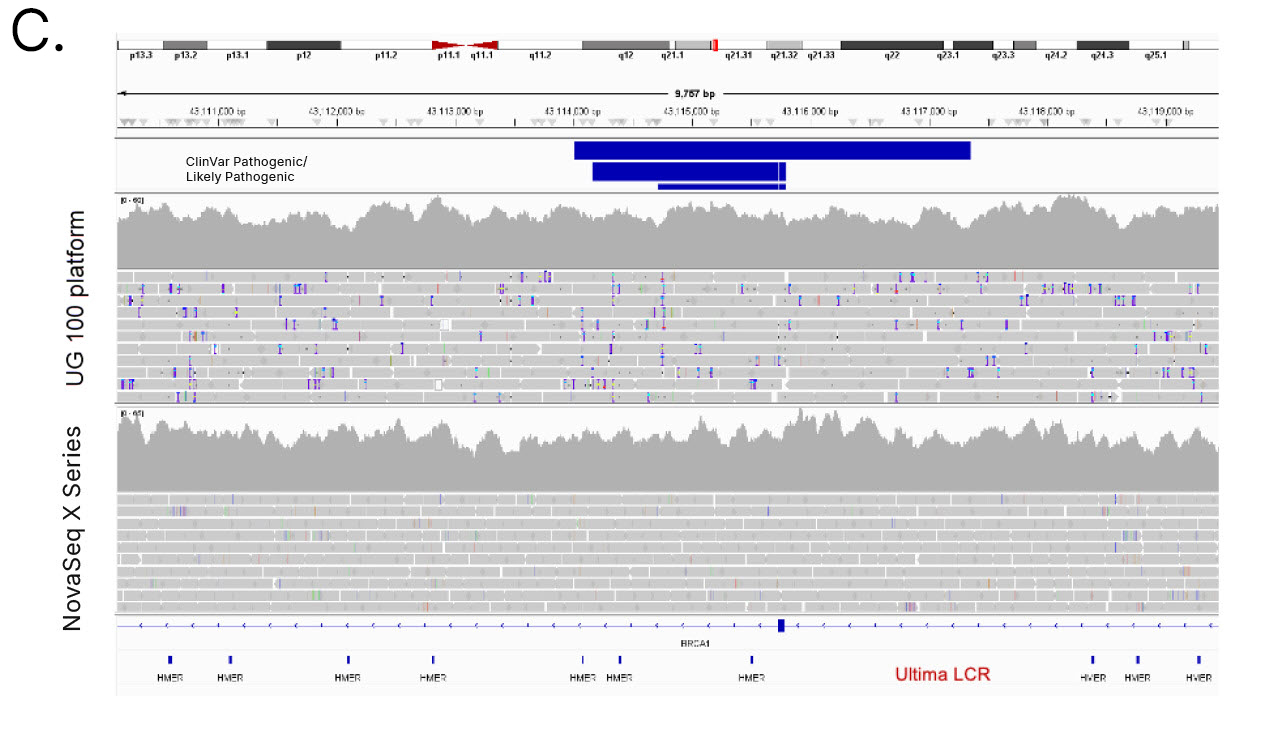
Figure 6: Loss of coverage with Ultima Genomics─Sequencing with the UG 100 platform results in loss of coverage in disease-relevant genes with high GC content, such as (A) B3GALT6 and (B) FMR1, and (C) significantly more indel calling errors in the BRCA1 gene compared to the NovaSeq X Series.
Summary
After evaluating Ultima Genomics and Illumina NGS platforms, the results demonstrate the superior performance of the NovaSeq X Series compared to the UG 100 platform for WGS. The NovaSeq X Series delivers higher accuracy and provides comprehensive coverage across the genome, including challenging regions. The performance of the UG 100 platform is inflated by the Ultima Genomics "high-confidence region," which excludes 4.2% of the genome from analysis where the platform’s performance is poor. The NovaSeq X Series generates the accuracy and genome coverage necessary to deliver biological insights into biologically relevant genes, whereas the UG 100 platform fails to accurately sequence genes with known associations to disease, such as B3GALT6 and BRCA1.
As established in this article, Ultima Genomics excludes various genomic regions from analysis to claim exceptional coverage and accuracy. The capability of the UG 100 platform to discover novel variants or even detect known variants in relevant genes is limited. In contrast, Illumina is a trusted global leader with 27 years of expertise and continues to provide comprehensive support and best-in-class product consistency, setting the standard for NGS solutions. The NovaSeq X Series and DRAGEN secondary analysis deliver accuracy and quality for comprehensive WGS at scale, without the compromises or tradeoffs required by Ultima Genomics technology.
Related links
Have a question about WGS?
Have questions about how Illumina platforms compare to Ultima Genomics? We can help you determine the total cost of ownership and recommend the best solution for your setup.
References
- Illumina. 25 greatest impacts in 25 years: Illumina and the evolution of genomics. illumina.com/company/news-center/feature-articles/25-greatest-impacts-in-25-years--a-look-back-at-illumina-and-the.html. Published April 3, 2023. Accessed July 22, 2025.
- Business Wire. Ultima announces UG 100 and reveals disruptive cost and accuracy profile to enable the era of the $100 genome and beyond. businesswire.com/news/home/20240206310974/en/Ultima-Announces-UG-100-and-Reveals-Disruptive-Cost-and-Accuracy-Profile-to-Enable-the-Era-of-the-%24100-Genome-and-Beyond. Published February 6, 2024. Accessed July 22, 2025.
- Daniels CA, Abdulkadir A, Cleveland MH, et al. A robust benchmark for detecting low-frequency variants in the HG002 Genome In A Bottle NIST reference material. Preprint. bioRxiv. 2024;2024.12.02.625685. Published 2024 Dec 5. doi:10.1101/2024.12.02.625685
- Wagner J, Olson ND, Harris L, et al. Benchmarking challenging small variants with linked and long reads. Cell Genom. 2022;2(5):100128. doi:10.1016/j.xgen.2022.100128
- Wagner J, Olson ND, Harris L, et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat Biotechnol. 2022;40(5):672-680. doi:10.1038/s41587-021-01158-1
- Illumina. Unlocking the full potential of Illumina genomes: The journey to enhanced variant calling quality with DRAGEN informatics and high-quality sequencing. https://www.illumina.com/science/genomics-research/articles/CMRG_hg38.html. Accessed July 17, 2025.
- Ultima Genomics. UG 100 Sequencing Platform specification sheet. Published 2025. Accessed July 17, 2025.
- Fotsing SF, Margoliash J, Wang C, et al. The impact of short tandem repeat variation on gene expression. Nat Genet. 2019;51(11):1652-1659. doi:10.1038/s41588-019-0521-9
- Van Damme T, Pang X, Guillemyn B, et al. Biallelic B3GALT6 mutations cause spondylodysplastic Ehlers-Danlos syndrome. Hum Mol Genet. 2018;27(20):3475-3487. doi:10.1093/hmg/ddy234
- Grigsby J. The fragile X mental retardation 1 gene (FMR1): historical perspective, phenotypes, mechanism, pathology, and epidemiology. Clin Neuropsychol. 2016;30(6):815-833. doi:10.1080/13854046.2016.1184652
- Semmler L, Reiter-Brennan C, Klein A. BRCA1 and Breast Cancer: a Review of the Underlying Mechanisms Resulting in the Tissue-Specific Tumorigenesis in Mutation Carriers. J Breast Cancer. 2019;22(1):1-14. doi:10.4048/jbc.2019.22.e6
- Fu X, Tan W, Song Q, Pei H, Li J. BRCA1 and Breast Cancer: Molecular Mechanisms and Therapeutic Strategies. Front Cell Dev Biol. 2022;10:813457. Published 2022 Mar 1. doi:10.3389/fcell.2022.813457
- Schulte J, Littleton JT. The biological function of the Huntingtin protein and its relevance to Huntington's Disease pathology. Curr Trends Neurol. 2011;5:65-78.
- Bishara J, Keens TG, Perez IA. The genetics of congenital central hypoventilation syndrome: clinical implications. Appl Clin Genet. 2018;11:135-144. Published 2018 Nov 15. doi:10.2147/TACG.S140629
- Coffin SL, Durham MA, Nitschke L, et al. Disruption of the ATXN1-CIC complex reveals the role of additional nuclear ATXN1 interactors in spinocerebellar ataxia type 1. Neuron. 2023;111(4):481-492.e8. doi:10.1016/j.neuron.2022.11.016
- Goswami R, Bello AI, Bean J, et al. The Molecular Basis of Spinocerebellar Ataxia Type 7. Front Neurosci. 2022;16:818757. Published 2022 Mar 24. doi:10.3389/fnins.2022.818757
- Leardini D, Messelodi D, Muratore E, et al. Role of CBL Mutations in Cancer and Non-Malignant Phenotype. Cancers (Basel). 2022;14(3):839. Published 2022 Feb 8. doi:10.3390/cancers14030839
- Winnepenninckx B, Debacker K, Ramsay J, et al. CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am J Hum Genet. 2007;80(2):221-231. doi:10.1086/510800
- Barratt KS, Arkell RM. ZIC2 in Holoprosencephaly. Adv Exp Med Biol. 2018;1046:269-299. doi:10.1007/978-981-10-7311-3_14
- Malerba A, Klein P, Lu-Nguyen N, et al. Established PABPN1 intranuclear inclusions in OPMD muscle can be efficiently reversed by AAV-mediated knockdown and replacement of mutant expanded PABPN1. Hum Mol Genet. 2019;28(19):3301-3308. doi:10.1093/hmg/ddz167
- Tazelaar GHP, Dekker AM, van Vugt JJFA, et al. Association of NIPA1 repeat expansions with amyotrophic lateral sclerosis in a large international cohort. Neurobiol Aging. 2019;74:234.e9-234.e15. doi:10.1016/j.neurobiolaging.2018.09.012
- Bavassano C, Eigentler A, Stanika R, et al. Bicistronic CACNA1A Gene Expression in Neurons Derived from Spinocerebellar Ataxia Type 6 Patient-Induced Pluripotent Stem Cells. Stem Cells Dev. 2017;26(22):1612-1625. doi:10.1089/scd.2017.0085
- Deng J, Yu J, Li P, et al. Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am J Hum Genet. 2020;106(6):793-804. doi:10.1016/j.ajhg.2020.04.011
- van Kuilenburg ABP, Tarailo-Graovac M, Richmond PA, et al. Glutaminase Deficiency Caused by Short Tandem Repeat Expansion in GLS. N Engl J Med. 2019;380(15):1433-1441. doi:10.1056/NEJMoa1806627
- Kühnel T, Leitão E, Lunzer R, et al. Repeat Expansions with Small TTTCA Insertions in MARCHF6 Cause Familial Myoclonus without Epilepsy. Mov Disord. 2025;40(7):1401-1408. doi:10.1002/mds.30192
- Goodman FR, Bacchelli C, Brady AF, et al. Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am J Hum Genet. 2000;67(1):197-202. doi:10.1086/302961
- Kumutpongpanich T, Ogasawara M, Ozaki A, et al. Clinicopathologic Features of Oculopharyngodistal Myopathy With LRP12 CGG Repeat Expansions Compared With Other Oculopharyngodistal Myopathy Subtypes. JAMA Neurol. 2021;78(7):853-863. doi:10.1001/jamaneurol.2021.1509
- Yu H, Wu J, Cong J, et al. Congenital insensitivity to pain associated with PRDM12 mutation: Two case reports and a literature review. Front Genet. 2023;14:1139161. Published 2023 Mar 20. doi:10.3389/fgene.2023.1139161